NEJM
Perspective
|
Beverly S. Mitchell, M.D.
This is an exciting time for cancer therapeutics. The identification of promising molecular targets has led to the development of many exciting new drugs for which an antitumor mechanism of action has been clearly delineated. Given the recent major advances in our understanding of the biology of cancer cells, one might surmise that an era of truly rational therapeutics has arrived. Nevertheless, we continue to find new therapeutic agents that target unforeseen molecular pathways.
The report by Richardson et al. in this issue of the Journal (pages 2609�2617) brings to the fore a relatively new and unexpected target, the 26S proteasome. This large, multi-subunit protein complex, which is present in high amounts in both the cytoplasm and nucleus of all eukaryotic cells, has the task of eliminating cellular proteins, including proteins that have been tagged for degradation through a complex modification termed "polyubiquitination." Proteins entering the proteasome are stripped of their ubiquitin, unfolded, and subsequently degraded through catalytic activities within the core of the proteasome. Substrates for ubiquitination and proteolytic degradation include a variety of proteins with such critical functions as regulation of the cell cycle, transcription, and apoptosis, as well as the regulation of chemotaxis, angiogenesis, and cell adhesion. The proteasome is thus an essential component of cellular metabolism.
At first glance, the features of the proteasome would scarcely make it a plausible target for highly selective cancer therapy. But the development of specific and potent chemical inhibitors of the proteasome has sparked considerable excitement about the therapeutic potential of this class of drugs, not only in cancer but also in a variety of inflammatory and immune disorders. Unlike other proteases, the proteasome contains an active-site N-terminal threonine residue that can be targeted by pharmacophores linked to short peptides. (A pharmacophore is the group of atoms in the pharmacologically active site of a drug.) The compound bortezomib (PS-341) contains a boronate moiety linked to a dipeptide (see Figure) and has exceedingly high affinity, specificity, and selectivity for catalytic activity of the proteasome. Furthermore, its inhibitory effects are reversible, allowing the return of most proteasome activity by 72 hours after administration. Bortezomib induces apoptosis in a wide variety of cancer-cell lines and other transformed cells, yet it has relatively few toxic effects on normal cells. In addition, it has considerable efficacy as a single agent against human tumor xenografts and primary cultures of tumor cells from patients with multiple myeloma, lymphoma, chronic lymphocytic leukemia, head and neck cancer, and prostate cancer. Even more exciting, bortezomib markedly enhances the apoptotic effects of irinotecan, gemcitabine, doxorubicin, and ionizing irradiation.
|
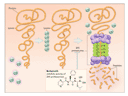
Why the selectivity and broad spectrum of activity? There is no single answer, but much research concerns the effects of proteasome inhibition on nuclear factor-
Modification of I![]() B in a way that
prevents its phosphorylation and ubiquitination results not only in
the retention of NF-
B in a way that
prevents its phosphorylation and ubiquitination results not only in
the retention of NF-![]() B in
the cytoplasm, but also in the sensitization of tumor cells to
chemotherapeutic agents and radiation. This result is evidence that
nuclear NF-
B in
the cytoplasm, but also in the sensitization of tumor cells to
chemotherapeutic agents and radiation. This result is evidence that
nuclear NF-![]() B inhibits cell death.
Since NF-
B inhibits cell death.
Since NF-![]() B could play a
part in the resistance of cancer cells to chemotherapy or irradiation,
it may be a therapeutic target in its own right or indirectly,
through specific inhibitors of I
B could play a
part in the resistance of cancer cells to chemotherapy or irradiation,
it may be a therapeutic target in its own right or indirectly,
through specific inhibitors of I![]() B.
As should be expected, however, the inhibition of the proteasome has
other consequences, including decreased activation of the mitogen-activated
protein-kinase pathway and up-regulation of p53 and the cell-cycle inhibitor
p27. Sorting out which effects contribute to the induction of cell
death in specific types of tumors and to the sensitization to drugs
and irradiation will occupy us for the foreseeable future.
B.
As should be expected, however, the inhibition of the proteasome has
other consequences, including decreased activation of the mitogen-activated
protein-kinase pathway and up-regulation of p53 and the cell-cycle inhibitor
p27. Sorting out which effects contribute to the induction of cell
death in specific types of tumors and to the sensitization to drugs
and irradiation will occupy us for the foreseeable future.
The clinical application of bortezomib has come about rapidly. A phase 1 trial involving 43 patients with solid tumors that were refractory to multiple treatments demonstrated some gastrointestinal toxicity and sensorineural toxicity but no dose-limiting hematologic toxicity.1 In another phase 1 trial involving patients with refractory hematologic cancers, the drug was well tolerated; of nine patients with multiple myeloma who could be evaluated, one had a complete response to therapy, and the other eight had a reduction in paraprotein levels, plasma cells in the marrow, or both.2 Toxic effects included thrombocytopenia and neutropenia. The phase 2 study by Richardson et al. presents compelling evidence that bortezomib has a role in the treatment of multiple myeloma. In patients with highly refractory disease, there was a 35 percent overall response rate, including a 10 percent rate of complete response, as defined by a normal serum protein concentration on electrophoresis. Toxic effects included thrombocytopenia, peripheral neuropathy, and neutropenia, but severe toxic effects were uncommon. On the basis of these studies, the Food and Drug Administration has very recently approved bortezomib for patients with refractory multiple myeloma.
The story is not over. There are currently ongoing phase 1 and 2 trials of bortezomib alone or in combination with other drugs for both hematologic cancers and solid tumors, and an international phase 3 trial is comparing bortezomib with dexamethasone in the treatment of myeloma. It would appear that the sensitization of tumor cells to cytotoxic drugs through inhibition of the proteasome will continue to generate enthusiasm for numerous clinical applications. A new "irrational" molecular target has come into its own.
Source Information
From the Division of Hematology/Oncology and the Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill.
References
- Aghajanian C, Soignet S, Dizon DS, et al. A phase I trial of the
novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin
Cancer Res 2002;8:2505-2511.
[Abstract/Full Text] - Orlowski RZ, Stinchcombe TE, Mitchell BS, et al. Phase I trial
of the proteasome inhibitor PS-341 in patients with refractory hematologic
malignancies. J Clin Oncol 2002;20:4420-4427.
[Abstract/Full Text]