NEJM
Genomic Medicine
|
Elizabeth G. Nabel, M.D.
Cardiovascular disease, including stroke, is the leading cause of illness and death in the United States. There are an estimated 62 million people with cardiovascular disease and 50 million people with hypertension in this country.1 In 2000, approximately 946,000 deaths were attributable to cardiovascular disease, accounting for 39 percent of all deaths in the United States.2 Epidemiologic studies and randomized clinical trials have provided compelling evidence that coronary heart disease is largely preventable.3 However, there is also reason to believe that there is a heritable component to the disease. In this review, I highlight what we know now about genetic factors in cardiovascular disease. As future genomic discoveries are translated to the care of patients with cardiovascular disease, it is likely that what we can do will change.
Lessons Learned from Monogenic Cardiovascular Disorders
Our understanding of the mechanism by which single genes can cause disease, even though such mechanisms are uncommon, has led to an understanding of the pathophysiological basis of more common cardiovascular diseases, which clearly are genetically complex. This point can be illustrated by a description of the genetic basis of specific diseases.
Elevated Levels of Low-Density Lipoprotein Cholesterol and Coronary Artery Disease
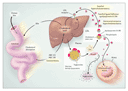
Low-density lipoprotein (LDL) is the major cholesterol-carrying lipoprotein in plasma and is the causal agent in many forms of coronary heart disease (Figure 1). Four monogenic diseases elevate plasma levels of LDL by impairing the activity of hepatic LDL receptors, which normally clear LDL from the plasma (Table 1). Familial hypercholesterolemia was the first monogenic disorder shown to cause elevated plasma cholesterol levels. The primary defect in familial hypercholesterolemia is a deficit of LDL receptors, and more than 600 mutations in the LDLR gene have been identified in patients with this disorder.5 One in 500 people is heterozygous for at least one such mutation, whereas only 1 in a million is homozygous at a single locus. Those who are heterozygous produce half the normal number of LDL receptors, leading to an increase in plasma LDL levels by a factor of 2 or 3, whereas LDL levels in those who are homozygous are 6 to 10 times normal levels. Homozygous persons have severe coronary atherosclerosis and usually die in childhood from myocardial infarction.
|
|
Deficiency of lipoprotein transport abolishes transporter activity, resulting in elevated cholesterol absorption and LDL synthesis. For example, mutations in the APOB-100 gene, which encodes apolipoprotein B-100, reduce the binding of apolipoprotein B-100 to LDL receptors and slow the clearance of plasma LDL, causing a disorder known as familial ligand-defective apolipoprotein B-100.6 One in 1000 people is heterozygous for one of these mutations; lipid profiles and clinical disease in such persons are similar to those of persons heterozygous for a mutation causing familial hypercholesterolemia.
Sitosterolemia, a rare autosomal disorder, results from loss-of-function mutations in genes encoding two ATP-binding cassette (ABC) transporters, ABC G5 and ABC G8,7,8 which act in concert to export cholesterol into the intestinal lumen, thereby diminishing cholesterol absorption. Autosomal recessive hypercholesterolemia is extremely rare (prevalence, <1 case per 10 million persons). The molecular cause is the presence of defects in a putative hepatic adaptor protein, which then fails to clear plasma LDL with LDL receptors.9 Mutations in the gene encoding that protein (ARH) elevate plasma LDL to levels similar to those seen in homozygous familial hypercholesterolemia.
In the majority of people with hypercholesterolemia in the general public, the condition is attributable to high-fat diets and poorly understood susceptibility and modifier genes. Study of the monogenic disorders, noted above, that disrupt LDL-receptor pathways has clarified the importance of cholesterol synthesis and excretion pathways in the liver and has highlighted molecular targets for regulating plasma cholesterol levels. For example, statin therapy for hypercholesterolemia is based on an understanding of the molecular basis of that disorder.
Hypertension
Hypertension is the most common disease in industrialized nations, with a prevalence above 20 percent in the general population. It imparts an increased risk of stroke, myocardial infarction, heart failure, and renal failure; many clinical trials have shown that reductions in blood pressure reduce the incidence of stroke and myocardial infarction.10 Multiple environmental and genetic determinants complicate the study of blood-pressure variations in the general population. In contrast, the investigation of rare mendelian forms of blood-pressure variation in which mutations in single genes cause marked extremes in blood pressure has been very informative (Table 2). These mutations, which impair renal salt handling, provide a molecular basis for understanding the pathogenesis of hypertension (Figure 2).11
|
|
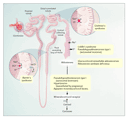
Investigation of families with severe hypertension or hypotension has identified mutations in genes that regulate these pathways. Pseudohypoaldosteronism type II is an autosomal dominant disorder characterized by hypertension, hyperkalemia, increased renal salt reabsorption, and impaired potassium- and hydrogen-ion excretion. Wilson and colleagues identified two genes causing pseudohypoaldosteronism type II; both encode proteins in the WNK family of serine�threonine kinases.12 Mutations in WNK1 are intronic deletions on chromosome 12p. Missense mutations in hWNK4, on chromosome 17, also cause pseudohypoaldosteronism type II. Immunofluorescence assays have shown that the proteins localize to distal nephrons and may serve to increase transcellular chloride conductance in the collecting ducts, leading to salt reabsorption, increased intravascular volume, and diminished secretion of potassium and hydrogen ions.
Abnormalities in the activity of aldosterone synthase produce hypertension
or hypotension. Glucocorticoid-remediable aldosteronism is an
autosomal dominant trait featuring early-onset hypertension with
suppressed renin activity and normal or elevated aldosterone levels.
This form of aldosteronism is caused by gene duplication arising from
an unequal crossover between two genes that encode enzymes in the
adrenal-steroid biosynthesis pathway (aldosterone synthase and 11![]() -hydroxylase).13,14
The chimeric gene encodes a protein with aldosterone synthase
activity that is ectopically expressed in the adrenal fasciculata
under the control of corticotropin rather than angiotensin II. Normal
cortisol production leads to constitutive aldosterone secretion,
plasma-volume expansion, hypertension, and suppressed renin levels.
Mutations that cause a loss of aldosterone synthase activity impair
renal salt retention and the secretion of potassium and hydrogen ions
in the distal nephrons and lead to severe hypotension as a result of
reduced intravascular volume.15
-hydroxylase).13,14
The chimeric gene encodes a protein with aldosterone synthase
activity that is ectopically expressed in the adrenal fasciculata
under the control of corticotropin rather than angiotensin II. Normal
cortisol production leads to constitutive aldosterone secretion,
plasma-volume expansion, hypertension, and suppressed renin levels.
Mutations that cause a loss of aldosterone synthase activity impair
renal salt retention and the secretion of potassium and hydrogen ions
in the distal nephrons and lead to severe hypotension as a result of
reduced intravascular volume.15
Mutations that alter renal ion channels and transporters give rise to Liddle's, Gitelman's, and Bartter's syndromes. Liddle's syndrome is an autosomal dominant trait characterized by early-onset hypertension, hypokalemic alkalosis, suppressed renin activity, and low plasma aldosterone levels due to mutations in the epithelial sodium channel.16,17 Loss-of-function mutations in the gene encoding the thiazide-sensitive sodium�chloride cotransporter in the distal convoluted tubules cause Gitelman's syndrome.18 Patients present in adolescence or early adulthood with neuromuscular signs and symptoms, a lower than normal blood pressure, a low serum magnesium level, and a low urinary calcium level. Bartter's syndrome can be produced by mutations in any of three genes required for normal salt reabsorption in the thick ascending loop of Henle; it can be distinguished from Gitelman's syndrome because it features increased urinary calcium levels and normal or reduced magnesium levels.19 In these inherited disorders, the net salt balance consistently predicts the blood pressure. As a result, new targets for antihypertensive therapy, including the epithelial sodium channel, other ion channels, and the WNK kinases, have been identified.
Thrombosis and Hemostasis
The blood-clotting system requires precise control of factors within and outside the coagulation cascade to prevent fatal bleeding or unwanted thrombosis. A common variant in the factor V gene, one encoding the substitution of glutamine for arginine at position 506 (Arg506Gln), prevents the degradation of factor V and promotes clot formation. This substitution, also known as factor V Leiden, has an allele frequency of 2 to 7 percent in European populations and has been observed in 20 to 50 percent of patients with venous thromboembolic disease.20,21,22 Factor V Leiden has incomplete penetrance and variable expression. Approximately 80 percent of persons who are homozygous for the mutation and 10 percent of those who are heterozygous will have thrombosis at some point in their lifetime.23,24 Factor V Leiden increases the risk of myocardial infarction, stroke, and venous thrombosis in men.25 In a subgroup of patients, thrombosis is associated with coinheritance of gene mutations that modify the factor V Leiden phenotype.26,27,28,29 Identification of gene modifiers is an area of active research and is essential for distinguishing, among persons who are heterozygous for factor V Leiden, the 10 percent in whom serious thrombosis will develop from the 90 percent who will have no symptoms.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy is the most common monogenic cardiac
disorder and the most frequent cause of sudden death from cardiac causes
in children and adolescents.30 On
the basis of the evaluation of echocardiograms from a large
population of young persons, the incidence of hypertrophic
cardiomyopathy has been estimated at approximately 1 in 500 persons.31
Hypertrophic cardiomyopathy is transmitted in an autosomal dominant
pattern. Mutations in the genes encoding proteins of the
myocardial-contractile apparatus cause the disease (Figure
3).32 Investigators have found multiple causative
mutations in at least 10 different sarcomeric proteins,33
including cardiac ![]() -myosin
heavy chain, cardiac myosin-binding protein, cardiac troponin T,
cardiac troponin I,
-myosin
heavy chain, cardiac myosin-binding protein, cardiac troponin T,
cardiac troponin I, ![]() -tropomyosin,
essential and regulatory light chains, and cardiac actin.
-tropomyosin,
essential and regulatory light chains, and cardiac actin.
|
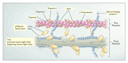
The pathologic features of hypertrophic cardiomyopathy consist of marked left ventricular hypertrophy, a thickened ventricular septum, atrial enlargement, and a small left ventricular cavity. Hypertrophy and disarray of the myocytes and interstitial fibrosis are present throughout the myocardium. The cardiac phenotype and clinical course of patients with hypertrophic cardiomyopathy are highly variable with regard to the pattern and degree of hypertrophy, the age at onset, and the clinical outcome. This variability is due partly to the different functions performed by mutant sarcomeric proteins. For example, a mutation in the gene encoding
Many of the mutations affecting ![]() -myosin
heavy chain involve the head and head�rod junction of the heavy
chain (Figure 3); some of these lead to pathologic
changes early in life and produce severe hypertrophy. The clinical
course varies even among persons with these mutations; an arginine-to-glutamine
substitution at position 403 (Arg403Gln) and an arginine-to-tryptophan
substitution at position 719 (Arg719Trp) predispose persons to
sudden death and heart failure, whereas a phenylalanine-to-cysteine substitution
at position 513 (Phe513Cys), a leucine-to-valine substitution at
position 908 (Leu908Val), and a glycine-to-glutamic acid substitution
at position 256 (Gly256Glu) cause less severe clinical disease.30,36
In contrast, mutations affecting cardiac myosin-binding protein
produce late-onset hypertrophic cardiomyopathy and are associated
with a more favorable prognosis.37
-myosin
heavy chain involve the head and head�rod junction of the heavy
chain (Figure 3); some of these lead to pathologic
changes early in life and produce severe hypertrophy. The clinical
course varies even among persons with these mutations; an arginine-to-glutamine
substitution at position 403 (Arg403Gln) and an arginine-to-tryptophan
substitution at position 719 (Arg719Trp) predispose persons to
sudden death and heart failure, whereas a phenylalanine-to-cysteine substitution
at position 513 (Phe513Cys), a leucine-to-valine substitution at
position 908 (Leu908Val), and a glycine-to-glutamic acid substitution
at position 256 (Gly256Glu) cause less severe clinical disease.30,36
In contrast, mutations affecting cardiac myosin-binding protein
produce late-onset hypertrophic cardiomyopathy and are associated
with a more favorable prognosis.37
Numerous factors other than sarcomere mutations determine the pathologic features and clinical course of hypertrophic cardiomyopathy. The identical sarcomere mutation can cause different hypertrophic changes and clinical outcomes among kindreds, even within the same pedigree.38,39 Gene modifiers, the environment, sex, and acquired conditions (such as ischemic or valvular heart disease) may account for these differences. Studies examining polymorphisms in the genes encoding angiotensin II, aldosterone, and endothelin that may modify the phenotype of hypertrophic cardiomyopathy have not yielded consistent results.40,41,42,43 Interestingly, clinically affected persons with two mutations in the same gene or different genes (compound heterozygotes) have also been described.44
Cardiac Arrhythmias
In 2001, about 450,000 people in the United States died suddenly from
cardiac arrhythmias.1,2
Genetic factors may modify the risk of arrhythmia in the setting of
common environmental risks. Arrhythmia-susceptibility genes have been
identified and provide insight into the molecular pathogenesis of
lethal and nonlethal arrhythmias (Table 3).45
The SCN5A gene encodes ![]() subunits that form the sodium channels responsible for initiating
cardiac action potentials.46 Mutations
in SCN5A cause several familial forms of arrhythmias,
including the long-QT syndrome, idiopathic ventricular fibrillation,
and cardiac-conduction disease.47,48,49,50,51
A recently identified variant of the SCN5A gene, one with a transversion
of cytosine to adenine in codon 1102, causing a serine-to-tyrosine
substitution at position 1102 (S1102Y), has been associated with
arrhythmia in black Americans.52 The variant allele
(Y1102) accelerates sodium-channel activation and increases the
likelihood of abnormal cardiac repolarization and arrhythmia. About
13 percent of blacks carry one Y1102 allele,52
which does not cause arrhythmia in most carriers. However, studies
such as this point to the usefulness of molecular markers for the
prediction of susceptibility to arrhythmia in persons with acquired or
other genetic risk factors.
subunits that form the sodium channels responsible for initiating
cardiac action potentials.46 Mutations
in SCN5A cause several familial forms of arrhythmias,
including the long-QT syndrome, idiopathic ventricular fibrillation,
and cardiac-conduction disease.47,48,49,50,51
A recently identified variant of the SCN5A gene, one with a transversion
of cytosine to adenine in codon 1102, causing a serine-to-tyrosine
substitution at position 1102 (S1102Y), has been associated with
arrhythmia in black Americans.52 The variant allele
(Y1102) accelerates sodium-channel activation and increases the
likelihood of abnormal cardiac repolarization and arrhythmia. About
13 percent of blacks carry one Y1102 allele,52
which does not cause arrhythmia in most carriers. However, studies
such as this point to the usefulness of molecular markers for the
prediction of susceptibility to arrhythmia in persons with acquired or
other genetic risk factors.
|
The HERG gene encodes
Analysis of Complex Cardiovascular Traits
Although many single genes have been identified as the basis of monogenic cardiovascular disorders, fewer genes underlying common complex cardiovascular diseases have been identified.58 Multiple risk factors, gene�environment interactions, and an absence of rough estimates of the number of genes that influence a single trait all complicate study design. Current research on complex cardiovascular traits focuses on the identification of genetic variants that enhance the susceptibility to given conditions.
Gene Polymorphisms
Association studies provide a powerful approach to identifying DNA variants underlying complex cardiovascular traits and are very useful for narrowing a candidate interval identified by linkage analysis. Improved genotyping techniques, such as genome-wide scanning of single-nucleotide polymorphisms59,60,61 and mapping of single-nucleotide polymorphisms identifying common haplotypes in the human genome, are facilitating association studies of loci spanning the entire genome. This point can be illustrated by recent examples of case�control studies that used high-throughput genomic techniques to investigate genetic variants in a large number of candidate genes for myocardial infarction, premature coronary artery disease, and heart failure.
Polymorphism-association studies compare the prevalence of a genetic marker in unrelated people with a given disease to the prevalence in a control population. Polymorphism-association studies of cardiovascular disease should be interpreted with caution when biologic plausibility has not been determined or is not known. Single-nucleotide polymorphisms in linkage disequilibrium may be functionally important, or alternatively, the polymorphism may just be a marker for another, yet to be identified, disease-causing sequence variant.
To determine genetic variants in myocardial infarction, Yamada and colleagues examined the prevalence of 112 polymorphisms in 71 candidate genes in patients with myocardial infarction and control patients in Japan.62 The analysis revealed one statistically significant association in men (a cytosine-to-thymine polymorphism at nucleotide 1019 in the connexin 37 gene) and two in women (the replacement of four guanines with five guanines at position �668 [4G�668/5G] in the plasminogen-activator inhibitor type 1 gene and the replacement of five adenines with six adenines at position �1171 [5A�1171/6A] in the stromelysin-1 gene), suggesting that these single-nucleotide polymorphisms may confer susceptibility to myocardial infarction in this population.
The GeneQuest study investigated 62 candidate genes in patients and their siblings with premature myocardial infarction (men <45 years old and women <50 years old).63 In this study, a case�control approach comparing genomic sequences in 72 single-nucleotide polymorphisms between persons with premature coronary artery disease and members of a control population identified three variants in the genes encoding thrombospondin-4, thrombospondin-2, and thrombospondin-1 that showed a statistical association with premature coronary artery disease. The biologic mechanisms by which these variants in thrombospondin proteins may lead to early myocardial infarction have yet to be identified.
Small and colleagues described an association between two polymorphisms in
adrenergic-receptor genes and the risk of congestive heart failure in
black Americans.64 Genotyping at two loci �
one encoding a variant ![]() 2c-adrenergic
receptor (involving the deletion of four amino acids [
2c-adrenergic
receptor (involving the deletion of four amino acids [![]() 2cDel322�325])
and the other encoding a variant
2cDel322�325])
and the other encoding a variant ![]() 1-adrenergic
receptor (with a glycine at amino acid position 389 [
1-adrenergic
receptor (with a glycine at amino acid position 389 [![]() 1Arg389])
� was performed in patients with heart failure and in controls. The
1Arg389])
� was performed in patients with heart failure and in controls. The
![]() 2cDel322�325 variant,
when present alone, conferred some degree of risk, whereas the
2cDel322�325 variant,
when present alone, conferred some degree of risk, whereas the ![]() 1Arg389
variant alone did not. However, black patients who were homozygous
for both variants had a markedly increased incidence of heart
failure. The presence of the
1Arg389
variant alone did not. However, black patients who were homozygous
for both variants had a markedly increased incidence of heart
failure. The presence of the ![]() 2cDel322�325
variant is associated with norepinephrine release at cardiac sympathetic-nerve
synapses, and the presence of the
2cDel322�325
variant is associated with norepinephrine release at cardiac sympathetic-nerve
synapses, and the presence of the ![]() 1Arg389
variant may increase the sensitivity of cardiomyocytes to norepinephrine.
The findings of this study suggest that the
1Arg389
variant may increase the sensitivity of cardiomyocytes to norepinephrine.
The findings of this study suggest that the ![]() 2cDel322�325
and
2cDel322�325
and ![]() 1Arg389
receptors act synergistically in blacks to increase the risk of heart
failure. Genotyping at these two loci may identify persons at risk
for the development or progression of heart failure and may predict
their response to therapy.
1Arg389
receptors act synergistically in blacks to increase the risk of heart
failure. Genotyping at these two loci may identify persons at risk
for the development or progression of heart failure and may predict
their response to therapy.
These studies highlight the importance of cardiovascular genotyping to
establish a molecular diagnosis, to stratify patients according to
risk, and especially to guide therapy. The field of pharmacogenetics �
that is, the use of genome-wide approaches to determine the role of
genetic variants in individual responses to drugs � has provided
data showing that genetic polymorphisms of proteins involved in drug
metabolism, transporters, and targets have important effects on the
efficacy of cardiovascular drugs65,66
(Table 4). For example, sequence variants in the ADRB2
gene, which encodes the ![]() 2-adrenergic
receptor, influence the response to
2-adrenergic
receptor, influence the response to ![]() 2-agonist
drugs.79,87
Two common polymorphisms of the receptor, an arginine-to-glycine
substitution at codon 16 (Gly16) and a glycine-to-glutamine
substitution at codon 27 (Glu27), are associated with increased
agonist-induced desensitization and increased resistance to
desensitization, respectively. There is marked linkage disequilibrium
between the polymorphisms at codons 16 and 27, with the result that
persons who are homozygous for Glu27 are also likely to be homozygous
for Gly16, whereas those who are homozygous for Gly16 may be
homozygous for Gln27 or Glu27 or heterozygous at codon 27.
2-agonist
drugs.79,87
Two common polymorphisms of the receptor, an arginine-to-glycine
substitution at codon 16 (Gly16) and a glycine-to-glutamine
substitution at codon 27 (Glu27), are associated with increased
agonist-induced desensitization and increased resistance to
desensitization, respectively. There is marked linkage disequilibrium
between the polymorphisms at codons 16 and 27, with the result that
persons who are homozygous for Glu27 are also likely to be homozygous
for Gly16, whereas those who are homozygous for Gly16 may be
homozygous for Gln27 or Glu27 or heterozygous at codon 27.
|
In a study that examined the effects of agonist-induced desensitization in the vasculature mediated by these polymorphisms, the investigators found that persons who were homozygous for Arg16 had nearly complete desensitization, as determined by measures of venodilation in response to isoproterenol, in contrast to persons homozygous for Gly16 and regardless of the codon 27 status.79 Similarly, persons homozygous for Gln27 had higher maximal venodilation in response to isoproterenol than those homozygous for Glu27, regardless of their codon 16 status. These data demonstrate that polymorphisms of the
Gene-Expression Profiling
Functional genomics, which is the study of gene function by means of parallel measurements of expression within control and experimental genomes, commonly involves the use of microarrays and serial analysis of gene expression. Microarrays are artificially constructed grids of DNA in which each element of the grid acts as a probe for a specific RNA sequence; each grid holds a DNA sequence that is a reverse complement to the target RNA sequence. Measurements of gene expression by means of microarrays are useful tools to establish molecular diagnoses, dissect the pathophysiologic features of a disease, and predict patients' response to therapy.88 Microarray analyses have been used to define a role for proliferative and inflammatory genes in the development of restenosis after the placement of coronary-artery stents. Zohlnh�fer and colleagues identified clusters of differentially expressed genes from coronary-artery neointima and peripheral-blood cells from patients with restenosis, as compared with samples of normal coronary arteries.89,90 The up-regulation of genes with functions in cell proliferation, the synthesis of extracellular matrix, cell adhesion, and inflammatory responses were more abundant in the samples of neointima from the patients with restenosis than in the samples of normal coronary artery. Many genes were expressed to a similar extent in the neointimal tissues and the peripheral-blood cells from patients with restenosis, suggesting the possible use of peripheral-blood cells as a substitute in microarray studies when cardiovascular tissue is not available.
Considerations for Molecular and Clinical Diagnosis
Genetic diagnosis � that is, primary classification on the basis of the presence of a mutation, with subsequent stratification according to risk � is not widely available for the diagnosis of monogenic cardiovascular disorders. Today, physical examination and routine testing, such as echocardiography to detect hypertrophic cardiomyopathy or electrocardiographic analysis of the long-QT syndrome, establish clinical diagnoses.91 Genetic diagnoses are then made by research-oriented genotyping of selected pedigrees. Current initiatives focus on the natural history of monogenic disorders in large numbers of patients with specific mutations, in order to identify persons at high risk for cardiovascular events, asymptomatic carriers in whom pharmacologic interventions will retard or prevent disease, and nonaffected family members whose concern about their health can be addressed. With regard to complex traits in more common cardiovascular diseases, current research is identifying functionally significant variations in DNA sequences that can establish a molecular diagnosis and influence patients' outcome.
I am indebted to the members of my laboratory for their
helpful comments.
Source Information
From the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Md.
Address reprint requests to Dr. Nabel at the National Heart, Lung, and Blood Institute, Bldg. 10/8C103, 10 Center Dr., Bethesda, MD 20892, or at [email protected].
References
- NHLBI morbidity and mortality chartbook, 2002. Bethesda, Md.: National Heart, Lung, and Blood Institute, May 2002. (Accessed June 10, 2003, at http://www.nhlbi.nih.gov/resources/docs/cht-book.htm.)
- NHLBI fact book, fiscal year 2002. Bethesda, Md.: National Heart, Lung, and Blood Institute, February 2003. (Accessed June 10, 2003, at http://www.nhlbi.nih.gov/about/factpdf.htm.)
- Cooper R, Cutler J, Desvignes-Nickens P, et al. Trends and
disparities in coronary heart disease, stroke, and other cardiovascular
diseases in the United States: findings of the National Conference on
Cardiovascular Disease Prevention. Circulation 2000;102:3137-3147.
[Abstract/Full Text] - Goldstein JL, Brown MS. The cholesterol quartet. Science
2001;292:1310-1312.
[Full Text] - Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic & molecular bases of inherited disease. 8th ed. Vol. 2. New York: McGraw-Hill, 2001:2863-913.
- Kane JP, Havel RJ. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic & molecular bases of inherited disease. 8th ed. Vol. 2. New York: McGraw-Hill, 2001:2717-52.
- Berge KE, Tian H, Graf GA, et al. Accumulation of dietary
cholesterol in sitosterolemia caused by mutations in adjacent ABC
transporters. Science 2000;290:1771-1775.
[Abstract/Full Text] - Lee M-H, Lu K, Hazard S, et al. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet 2001;27:79-83. [CrossRef][ISI][Medline]
- Garcia CK, Wilund K, Arca M, et al. Autosomal recessive
hypercholesterolemia caused by mutations in a putative LDL receptor adaptor
protein. Science 2001;292:1394-1398.
[Abstract/Full Text] - ALLHAT Officers and Coordinators for the ALLHAT Collaborative
Research Group. Major outcomes in high-risk hypertensive patients randomized
to angiotensin-converting enzyme inhibitor or calcium channel blocker vs.
diuretic: the Hypertensive and Lipid-Lowering Treatment to Prevent Heart
Attack (ALLHAT). JAMA 2002;288:2981-2997. [Erratum, JAMA 2003;289:178.]
[Abstract/Full Text] - Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell 2001;104:545-556. [ISI][Medline]
- Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human
hypertension caused by mutations in WNK kinases. Science 2001;293:1107-1112.
[Abstract/Full Text] - Lifton RP, Dluhy RG, Powers M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992;355:262-265. [ISI][Medline]
- Lifton RP, Dluhy RG, Powers M, et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat Genet 1992;2:66-74. [ISI][Medline]
- Mitsuuchi Y, Kawamoto T, Naiki Y, et al. Congenitally defective aldosterone biosynthesis in humans: the involvement of point mutations of the P-450C18 gene (CYP11B2) in CMO II deficient patients. Biochem Biophys Res Commun 1992;182:974-979. [Erratum, Biochem Biophys Res Commun 1992;184:1529-30.] [ISI][Medline]
- Shimkets RA, Warnock DG, Bositis CM, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 1994;79:407-414. [ISI][Medline]
- Hansson JH, Schild L, Lu Y, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci U S A 1995;92:11495-11499. [Abstract]
- Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 1996;12:24-30. [ISI][Medline]
- Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 1996;13:183-188. [ISI][Medline]
- Svensson PJ, Dahlb�ck B. Resistance to activated protein C as
a basis for venous thrombosis. N Engl J Med 1994;330:517-522.
[Abstract/Full Text] - Griffin JH, Evatt B, Wideman C, Fern�ndez JA. Anticoagulant protein C pathway defective in majority of thrombophilic patients. Blood 1993;82:1989-1993. [Abstract]
- Koster T, Rosendaal FR, de Ronde H, Bri�t E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet 1993;342:1503-1506. [ISI][Medline]
- Greengard JS, Eichinger S, Griffin JH, Bauer KA. Variability of
thrombosis among homozygous siblings with resistance to activated protein C
due to an Arg
 Gln mutation in the
gene for factor V. N Engl J Med 1994;331:1559-1562.
Gln mutation in the
gene for factor V. N Engl J Med 1994;331:1559-1562.
[Full Text] - Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk
of thrombosis in patients homozygous for factor V Leiden (activated protein
C resistance). Blood 1995;85:1504-1508.
[Abstract/Full Text] - Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg
PR, Miletich JP. Mutation in the gene coding for coagulation factor V and
the risk of myocardial infarction, stroke, and venous thrombosis in
apparently healthy men. N Engl J Med 1995;332:912-917.
[Abstract/Full Text] - Koeleman BPC, Reitsma PH, Allaart CF, Bertina RM. Activated
protein C resistance as an additional risk factor for the thrombosis in
protein C-deficient families. Blood 1994;84:1031-1035.
[Abstract/Full Text] - Z�ller B, Berntsdotter A, Garcia de Frutos P, Dahlb�ck B.
Resistance to activated protein C as an additional genetic risk factor in
hereditary deficiency of protein S. Blood 1995;85:3518-3523.
[Abstract/Full Text] - Van Boven HH, Reitsma PH, Rosendaal FR, et al. Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 1996;75:417-421. [ISI][Medline]
- De Stefano V, Martinelli I, Mannucci PM, et al. The risk of recurrent venous thromboembolism among heterozygous carriers of the G20210A prothrombin gene mutation. Br J Haematol 2001;113:630-635. [CrossRef][ISI][Medline]
- Seidman CE, Seidman JG. Hypertrophic cardiomyopathy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic & molecular bases of inherited disease. 8th ed. Vol. 4. New York: McGraw-Hill, 2001:5433-50.
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild
DE. Prevalence of hypertrophic cardiomyopathy in a general population of
young adults: echocardiographic analysis of 4111 subjects in the CARDIA
Study: Coronary Artery Risk Development in (Young) Adults. Circulation
1995;92:785-789.
[Abstract/Full Text] - Kamisago M, Sharma SD, DePalma SR, et al. Mutations in
sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med
2000;343:1688-1696.
[Abstract/Full Text] - Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001;104:557-567. [ISI][Medline]
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990;62:999-1006. [ISI][Medline]
- NHLBI Program for Genomic Applications. Genomics of cardiovascular development, adaptation, and remodeling. Boston: Beth Israel Deaconess Medical Center, 2003. (Accessed June 10, 2003, at http://cardiogenomics.med.harvard.edu/project-detail?project_id=230#data.)
- Roberts R, Sigwart U. New concepts in hypertrophic
cardiomyopathy. Circulation 2001;104:2113-2116.
[Full Text] - Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in
the gene for cardiac myosin-binding protein C and late-onset familial
hypertrophic cardiomyopathy. N Engl J Med 1998;338:1248-1257.
[Abstract/Full Text] - Fananapazir L, Epstein N. Genotype-phenotype correlations in
hypertrophic cardiomyopathy: insights provided by comparisons of kindreds
with distinct and identical
 -myosin
heavy chain gene mutations. Circulation 1994;89:22-32.
[Abstract]
-myosin
heavy chain gene mutations. Circulation 1994;89:22-32.
[Abstract]
- Kimura A, Harada H, Park JE, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997;16:379-382. [ISI][Medline]
- Lechin M, Quinones MA, Omran A, et al. Angiotensin-I converting
enzyme genotypes and left ventricular hypertrophy in patients with
hypertrophic cardiomyopathy. Circulation 1995;92:1808-1812.
[Abstract/Full Text] - Tesson F, Dufour C, Moolman JC, et al. The influence of the angiotensin I converting enzyme genotype in familial hypertrophic cardiomyopathy varies with the disease gene mutation. J Mol Cell Cardiol 1997;29:831-838. [CrossRef][ISI][Medline]
- Osterop AP, Kofflard MJ, Sandkuijl LA, et al. AT1 receptor
A/C1166 polymorphism contributes to cardiac hypertrophy in subjects with
hypertrophic cardiomyopathy. Hypertension 1998;32:825-830.
[Abstract/Full Text] - Ortlepp JR, Vosberg HP, Reith S, et al. Genetic polymorphisms
in the renin-angiotensin-aldosterone system associated with expression of
left ventricular hypertrophy in hypertrophic cardiomyopathy: a study of five
polymorphic genes in a family with a disease causing mutation in the myosin
binding protein C gene. Heart 2002;87:270-275.
[Abstract/Full Text] - Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman
CE. Homozygous mutation in cardiac troponin T: implications for hypertrophic
cardiomyopathy. Circulation 2000;102:1950-1955.
[Abstract/Full Text] - Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001;104:569-580. [ISI][Medline]
- Gellens ME, George AL Jr, Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A 1992;89:554-558. [Abstract]
- Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805-811. [ISI][Medline]
- Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995;376:683-685. [ISI][Medline]
- Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293-296. [CrossRef][ISI][Medline]
- Schott JJ, Alshinawi C, Kyndt F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 1999;23:20-21. [CrossRef][ISI][Medline]
- Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 2001;409:1043-1047. [CrossRef][ISI][Medline]
- Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A
sodium channel implicated in risk of cardiac arrhythmia. Science
2002;297:1333-1336.
[Abstract/Full Text] - Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995;81:299-307. [ISI][Medline]
- Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999;97:175-187. [ISI][Medline]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996;384:78-80. [ISI][Medline]
- Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 1996;384:80-83. [ISI][Medline]
- Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in
long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation
2000;102:1178-1185.
[Abstract/Full Text] - Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie
complex traits. Science 2002;298:2345-2349.
[Abstract/Full Text] - Mohlke KL, Erdos MR, Scott LJ, et al. High-throughput screening
for evidence of association by using mass spectrometry genotyping on DNA
pools. Proc Natl Acad Sci U S A 2002;99:16928-16933.
[Abstract/Full Text] - Oliphant A, Barker DL, Stuelpnagel JR, Chee MS. BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques 2002;:56-8, 60.
- Cargill M, Altshuler D, Ireland J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 1999;22:231-238. [Erratum, Nat Genet 1999;23:373.] [CrossRef][ISI][Medline]
- Yamada Y, Izawa H, Ichihara S, et al. Prediction of the risk of
myocardial infarction from polymorphisms in candidate genes. N Engl J Med
2002;347:1916-1923.
[Abstract/Full Text] - Topol EJ, McCarthy J, Gabriel S, et al. Single nucleotide
polymorphisms in multiple novel thrombospondin genes may be associated with
familial premature myocardial infarction. Circulation 2001;104:2641-2644.
[Abstract/Full Text] - Small KM, Wagoner LE, Levin AM, Kardia SLR, Liggett SB.
Synergistic polymorphisms of 1-
and
 2c-adrenergic
receptors and the risk of congestive heart failure. N Engl J Med
2002;347:1135-1142.
2c-adrenergic
receptors and the risk of congestive heart failure. N Engl J Med
2002;347:1135-1142.
[Abstract/Full Text] - Weinshilboum R. Inheritance and drug response. N Engl J Med
2003;348:529-537.
[Full Text] - Evans WE, McLeod HL. Pharmacogenomics -- drug disposition, drug
targets, and side effects. N Engl J Med 2003;348:538-549.
[Full Text] - Hoffmeyer S, Burk O, von Richter O, et al. Functional
polymorphisms of the human multidrug-resistance gene: multiple sequence
variations and correlation of one allele with P-glycoprotein expression and
activity in vivo. Proc Natl Acad Sci U S A 2000;97:3473-3478.
[Abstract/Full Text] - Sakaeda T, Nakamura T, Horinouchi M, et al. MDR1 genotype-related pharmacokinetics of digoxin after single oral administration in healthy Japanese subjects. Pharm Res 2001;18:1400-1404. [CrossRef][ISI][Medline]
- Stavroulakis GA, Makris TK, Krespi PG, et al. Predicting response to chronic antihypertensive treatment with fosinopril: the role of angiotensin-converting enzyme gene polymorphism. Cardiovasc Drugs Ther 2000;14:427-432. [CrossRef][ISI][Medline]
- O'Toole L, Stewart M, Padfield P, Channer K. Effect of the insertion/deletion polymorphism of the angiotensin-converting enzyme gene on response to angiotensin-converting enzyme inhibitors in patients with heart failure. J Cardiovasc Pharmacol 1998;32:988-994. [CrossRef][ISI][Medline]
- Myerson SG, Montgomery HE, Whittingham M, et al. Left
ventricular hypertrophy with exercise and ACE gene insertion/deletion
polymorphism: a randomized controlled trial with losartan. Circulation
2001;103:226-230.
[Abstract/Full Text] - Kohno M, Yokokawa K, Minami M, et al. Association between angiotensin-converting enzyme gene polymorphisms and regression of left ventricular hypertrophy in patients treated with angiotensin-converting enzyme inhibitors. Am J Med 1999;106:544-549. [CrossRef][ISI][Medline]
- McNamara DM, Holubkov R, Janosko K, et al. Pharmacogenetic
interactions between -blocker
therapy and the angiotensin-converting enzyme deletion polymorphism in
patients with congestive heart failure. Circulation 2001;103:1644-1648.
[Abstract/Full Text] - Prasad A, Narayanan S, Husain S, et al. Insertion-deletion
polymorphism of the ACE gene modulates reversibility of endothelial
dysfunction with ACE inhibition. Circulation 2000;102:35-41.
[Abstract/Full Text] - Yoshida H, Mitarai T, Kawamura T, et al. Role of the deletion of polymorphism of the angiotensin converting enzyme gene in the progression and therapeutic responsiveness of IgA nephropathy. J Clin Invest 1995;96:2162-2169. [ISI][Medline]
- Perna A, Ruggenenti P, Testa A, et al. ACE genotype and ACE inhibitors induced renoprotection in chronic proteinuric nephropathies. Kidney Int 2000;57:274-281. [CrossRef][ISI][Medline]
- Penno G, Chaturvedi N, Talmud PJ, et al. Effect of angiotensin-converting enzyme (ACE) gene polymorphism on progression of renal disease and the influence of ACE inhibition in IDDM patients: findings from the EUCLID Randomized Controlled Trial: EURODIAB Controlled Trial of Lisinopril in IDDM. Diabetes 1998;47:1507-1511. [Abstract]
- Marian AJ, Safavi F, Ferlic L, Dunn JK, Gotto AM, Ballantyne CM. Interactions between angiotensin-I converting enzyme insertion/deletion polymorphism and re-sponse of plasma lipids and coronary atherosclerosis to treatment with fluvastatin: the Lipoprotein and Coronary Atherosclerosis Study. J Am Coll Cardiol 2000;35:89-95. [CrossRef][ISI][Medline]
- Dishy V, Sofowora GG, Xie H-G, et al. The effect of common
polymorphisms of the 2-adrenergic
receptor on agonist-mediated vascular desensitization. N Engl J Med
2001;345:1030-1035.
[Abstract/Full Text] - Drysdale CM, McGraw DW, Stack CB, et al. Complex promoter and
coding region beta 2-adrenergic receptor haplotypes alter receptor
expression and predict in vivo responsiveness. Proc Natl Acad Sci U S A
2000;97:10483-10488.
[Abstract/Full Text] - Cockcroft JR, Gazis AG, Cross DJ, et al. Beta-2 adrenoreceptor
polymorphism determines vascular reactivity in humans. Hypertension
2000;36:371-375.
[Abstract/Full Text] - Gerdes LU, Gerdes C, Kervinen K, et al. The apolipoprotein
epsilon4 allele determines prognosis and the effect on prognosis of
simvastatin in survivors of myocardial infarction: a substudy of the
Scandinavian Simvastatin Survival Study. Circulation 2000;101:1366-1371.
[Abstract/Full Text] - Pedro-Botet J, Schaefer EJ, Bakker-Arkema RG, et al. Apolipoprotein E genotype affects plasma lipid response to atorvastatin in a gender specific manner. Atherosclerosis 2001;158:183-193. [CrossRef][ISI][Medline]
- Ballantyne CM, Herd JA, Stein EA, et al. Apolipoprotein E genotypes and response of plasma lipids and progression-regression of coronary atherosclerosis to lipid-lowering drug therapy. J Am Coll Cardiol 2000;36:1572-1578. [CrossRef][ISI][Medline]
- Kuivenhoven JA, Jukema JW, Zwinderman AH, et al. The role of a
common variant of the cholesteryl ester transfer protein gene in the
progression of coronary atherosclerosis. N Engl J Med 1998;338:86-93.
[Abstract/Full Text] - Sesti F, Abbott GW, Wei J, et al. A common polymorphism
associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U
S A 2000;97:10613-10618.
[Abstract/Full Text] - Liggett SB. Beta(2)-adrenergic receptor pharmacogenetics. Am J
Respir Crit Care Med 2000;161:S197-S201.
[Full Text] - Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185-97.
- Zohlnh�fer D, Richter T, Neumann F-J, et al. Transcriptome
analysis reveals a role of interferon-
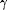 in human neointima formation. Mol Cell 2001;7:1059-1069.
[CrossRef][ISI][Medline]
in human neointima formation. Mol Cell 2001;7:1059-1069.
[CrossRef][ISI][Medline]
- Zohlnh�fer D, Klein CA, Richter T, et al. Gene expression
profiling of human stent-induced neointima by cDNA array analysis of
microscopic specimens retrieved by helix cutter atherectomy: detection of
FK506-binding protein 12 upregulation. Circulation 2001;103:1396-1402.
[Abstract/Full Text] - Maron BJ, Moller JH, Seidman CE, et al. Impact of laboratory
molecular diagnosis on contemporary diagnostic criteria for genetically
transmitted cardiovascular diseases: hypertrophic cardiomyopathy, long-QT
syndrome, and Marfan syndrome: a statement for healthcare professionals from
the councils on clinical cardiology, cardiovascular disease in the young,
and basic science, American Heart Association. Circulation
1998;98:1460-1471.
[Full Text]