NEJM
|
John M. Goldman, F.R.C.P., and Junia V. Melo, M.D., Ph.D.
Chronic myeloid leukemia (CML) was probably the first form of leukemia to be recognized as a distinct entity. In 1845, two patients were described as having massive splenomegaly associated with leukocytosis,1 which seemed to be a novel entity not explained by the other causes of splenomegaly, such as tuberculosis, that were already widely accepted in the 1840s. The first important clue to its pathogenesis came only very much later, when in 1960 newly developed techniques for studying human cells in mitosis allowed Nowell and Hungerford to detect a consistent chromosomal abnormality,2 later termed the Philadelphia (Ph1, or just Ph) chromosome and identified as 22q�, in persons with this disease. In 1973, Rowley observed that the Ph chromosome resulted from a reciprocal translocation that also involved chromosome 9; the abnormality is now designated t(9;22)(q34;q11).3 In the 1980s, the Ph chromosome was shown to carry a unique fusion gene, termed BCR-ABL,4 the generation of which is now believed to be the principal cause of the chronic phase of CML.
Until the 1980s, CML was regarded as incurable and thus inexorably fatal. We know now that selected patients can be treated, and in many cases cured, by allogeneic stem-cell transplantation. However, efforts to extend this form of treatment to all patients with CML have been thwarted by the lack of suitable donors and the increased incidence of potentially lethal graft-versus-host disease (GVHD) in older recipients. The recent introduction into clinical practice of a tyrosine kinase inhibitor that specifically blocks the enzymatic action of the BCR-ABL fusion protein promises to be a major contribution to the management of CML and may also prove to be the lead agent that ushers in an era of success with molecularly targeted therapy for other leukemias, lymphomas, and cancers.
The evolving story of CML and modern views on its biology and treatment have been reviewed in the Journal and elsewhere in recent years.5,6,7,8 Here, we briefly summarize recent advances in knowledge related to the pathogenesis of CML and indicate where these may have important therapeutic implications.
Cytokinetics
It is generally believed that CML develops when a single, pluripotential, hematopoietic stem cell acquires a Ph chromosome carrying the BCR-ABL fusion gene, which confers on its progeny a proliferative advantage over normal hematopoietic elements and thus allows the Ph-positive clone gradually to displace residual normal hematopoiesis.9,10 The evidence for this hypothesis derives in part from the consistency of the molecular abnormality in any given patient, but the mechanism by which the molecular and cytogenetic changes occur remains enigmatic. Similarly, the molecular basis of the apparent proliferative advantage is not well defined, but it may relate in part to constitutive expression by leukemic progenitors of growth-stimulating factors, notably interleukin-3 and granulocyte colony-stimulating factor.11,12 Moreover, CML cells seem to survive longer than their normal counterparts, as a result of a defective apoptotic response to stimuli that would otherwise lead to physiologic cell death.13
Whatever the primary mechanism of the disease, it is clear that the myeloid mass is greatly increased in patients with newly diagnosed disease, owing to an expansion of mature elements, as well as to increased numbers of progenitor cells and putative stem cells.10 The latter probably include a population of leukemic cells that are "deeply quiescent" and that may thus be relatively resistant to standard chemotherapy.14 Until the 1980s, there was uncertainty as to whether substantial numbers of normal stem cells were still present in the bone marrow (or elsewhere) in patients with a new diagnosis of CML. However, several findings � the demonstration of the presence of Ph-negative progenitors in myeloid-cell cultures,15 the observation that Ph-negative progenitor cells can be identified in the blood after high-dose chemotherapy,16 and the ability of interferon alfa to induce Ph-negativity in the marrow � all constitute persuasive circumstantial evidence that the Ph-positive clone displaces normal hematopoiesis but does not destroy residual normal stem cells. The final proof comes from the observation that imatinib mesylate, a new, ABL-specific tyrosine kinase inhibitor (formerly called STI571 and marketed under the name Gleevec in the United States and Glivec in Europe), can induce complete or nearly complete cytogenetic remissions in up to 80 percent of patients.17,18
It is reasonably certain that the majority of patients who survive allogeneic stem-cell transplantation and who appear to be free of disease five years after the procedure can be regarded as cured,19 but the definition and mechanism of "cure" remain controversial. In other words, cure might theoretically require the total eradication of all leukemia cells from a patient's body, but alternatively, an operational definition of cure might apply if low numbers of leukemia cells persisted but were unable to reestablish clinical disease. Such symbiosis of low numbers of leukemia cells with normal cells could, for example, be due to the continuing action of a graft-versus-leukemia effect after allogeneic stem-cell transplantation.20
Currently, the reverse-transcriptase polymerase chain reaction (RT-PCR) is the most sensitive method for detecting low numbers of BCR-ABL transcripts in a patient after apparently successful stem-cell transplantation.21 In practice, the results may be difficult to interpret because even if the test is negative, there may still be a million or more residual Ph-positive cells in the body; in other cases, the test may be persistently positive at a low level for many years. Moreover, it has been suggested that some residual Ph-positive cells may be "transcriptionally silent" and thus not detectable by conventional RT-PCR techniques,22 although this idea has been disputed.23,24 Finally, interpretation of RT-PCR results might theoretically be confounded by the fact that BCR-ABL transcripts can be detected at a very low level in the blood of many normal persons, the majority of whom will never have CML.25,26
Cytogenetics
The mechanism by which the Ph chromosome is first formed and the time required for progression to overt disease are unknown. Radiation may play a role in some cases, since persons exposed to high-dose irradiation have a significantly increased risk of leukemia,27 and high-dose irradiation of myeloid cell lines in vitro induces the expression of BCR-ABL transcripts indistinguishable from those that characterize CML.28 It has also been proposed that the close proximity of the BCR and ABL genes in hematopoietic cells in interphase may favor translocations between the two genes.29 Very recently, a 76-kb duplicon (a two-copy DNA repeat sequence) was identified on chromosome 9 near the ABL gene and on chromosome 22 near the BCR gene; it may be implicated in the translocation, but the mechanism is purely speculative.30
The BCR-ABL gene on the Ph chromosome is, of course, expressed in all patients with CML, but the ABL-BCR gene on the 9q+ derivative is expressed in only 70 percent of cases.31 Of great interest are the recent observations that about 20 percent of patients with CML have deletions of chromosomal material of varying size on the derivative 9q+ and that patients who have such deletions have significantly shorter survival than those who do not.32,33 These deletions presumably occur at the same time as the formation of the Ph chromosome, and their recognition will probably prove important in predicting survival for individual patients. Nonexpression of the ABL-BCR gene, which is always included in the deleted region, does not by itself have the same ominous prognostic implication.34 Thus, the identification of the precise gene or genes on 9q+, the deletion of which adversely influences prognosis in CML, would be an important achievement.
It is generally believed that the Ph-positive clone has an increased susceptibility to additional molecular changes that underlie disease progression. If so, the nonrandom cytogenetic changes identified in 60 to 80 percent of patients with disease in blastic transformation35 � notably +8, +Ph, +19, and i(17)q � should provide some clues as to the activation of oncogenes (other than BCR-ABL) or the deletion of tumor-suppressor genes in the transformation from chronic to advanced phase disease. However, in only a minority of cases has the transformation actually been linked to mutations, deletions, or altered expression of known genes, notably p53,36 p16,37 Rb,38 and EVI-1.39 In general, no specific pattern can be discerned. All in all, it seems likely that a variety of molecular mechanisms, rather than a single gene defect, underlies the arrest of maturation that occurs in a subclone of Ph-positive cells and manifests itself in the blastic phase of the disease. The advent of powerful microarray techniques that permit comparisons of gene-expression profiles should help to identify the genes involved in this transformation.
Although the majority of patients with a leukemia that appears on
morphologic grounds to be CML prove to have a Ph chromosome and a
classic BCR-ABL fusion gene, some do not. About one third of
the patients with CML who appear to have normal karyotypes actually
have a cytogenetically occult BCR-ABL gene, usually located on
a normal-appearing chromosome 22 but very occasionally on chromosome
9.40 In the remaining patients, in whom the
disease is described as Ph-negative, BCR-ABL�negative, the
leukemia has no known molecular basis. There is also a very small
group of patients with a form of chronic leukemia superficially
resembling classic CML who have consistent cytogenetic aberrations
(in most cases, translocations) other than the Ph chromosome (Table
1). The commonest non-Ph translocations are t(5;12)(q33;p13)41
and t(8;13) (p11;q12).42 Both translocations
generate fusion genes, one component of which is a gene that encodes
a receptor tyrosine kinase � namely, platelet-derived growth factor
receptor ![]() in t(5;12) and
fibroblast growth factor receptor 1 in t(8;13). Functional studies
performed with these fusion genes suggest that they cause a chronic
myeloid leukemia by signaling pathways that closely parallel the
pathways activated by the BCR-ABL oncoprotein.43,44,45
in t(5;12) and
fibroblast growth factor receptor 1 in t(8;13). Functional studies
performed with these fusion genes suggest that they cause a chronic
myeloid leukemia by signaling pathways that closely parallel the
pathways activated by the BCR-ABL oncoprotein.43,44,45
|
Molecular Events
The idea that CML, like other cancers, may be the result of a multistep pathogenetic process was first broached more than 20 years ago,46 but there is still very little evidence of any acquired molecular abnormalities preceding the t(9;22) translocation. Instead, it seems more likely that the generation of a classic BCR-ABL fusion gene in a specific type of cell (namely, a pluripotential hematopoietic stem cell), possibly under conditions of reduced immunologic surveillance, is sufficient to initiate the expansion of a hematopoietic clone that leads to CML. The proposition that the acquisition of a BCR-ABL fusion gene is the first step in the genesis of CML is supported by murine models in which a CML-like disease has been produced by transfecting stem cells with a BCR-ABL gene47,48,49; however, once established, the tempo or aggressiveness of the chronic phase disease varies from patient to patient and thus must be influenced by other factors.
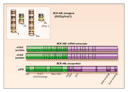
The classic BCR-ABL gene of CML results from the fusion of parts of two normal genes: the ABL gene on chromosome 9 and the BCR gene on chromosome 22. Both genes are ubiquitously expressed in normal tissues, but their precise functions are not well defined. In the translocation that forms the fusion gene, a break occurs in ABL somewhere upstream of exon a2, and simultaneously a break occurs in the major breakpoint cluster region of the BCR gene. As a result, a 5' portion of BCR and a 3' portion of ABL are juxtaposed on a shortened chromosome 22 (the derivative 22q�, or Ph, chromosome) (Figure 1). The messenger RNA (mRNA) molecules transcribed from this hybrid gene usually contain one of two BCR-ABL junctions, designated e13a2 (formerly b2a2) and e14a2 (or b3a2). There is no evidence that the type of junction has any prognostic significance. Both BCR-ABL mRNA molecules are translated into an 210-kD oncoprotein, usually referred to as p210BCR-ABL. Other variant breakpoints and fusions can give rise to full-length, functionally oncogenic BCR-ABL proteins, notably p190BCR-ABL (associated with an e1a2 mRNA junction) and p230BCR-ABL (associated with an e19a2 mRNA junction),50 but they are rather rare in classic CML.51,52
|
The leukemogenic potential of p210BCR-ABL resides in the fact that the normally regulated tyrosine kinase activity of the ABL protein is constitutively activated by the juxtaposition of alien BCR sequences (Figure 2). BCR acts by promoting dimerization of the oncoprotein, such that the two adjacent BCR-ABL molecules phosphorylate each other on tyrosine residues in their kinase-activation loops.56,57 The uncontrolled kinase activity of BCR-ABL then usurps the physiologic functions of the normal ABL enzyme by interacting with a variety of effector proteins, the net result of which is deregulated cellular proliferation, decreased adherence of leukemia cells to the bone marrow stroma, and a reduced apoptotic response to mutagenic stimuli. Unfortunately, the relative contributions of these effects to the phenotype of chronic-phase CML is still poorly understood.8
|
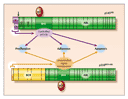
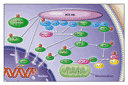
The structure of the BCR-ABL protein and the biochemical pathways in which it is involved have been extensively studied.58 Knowledge of the roles of several functional domains derived from the parental BCR and ABL proteins allows one to test for certain properties of the fusion product (Figure 1 and Figure 2). Thus, the tyrosine kinase encoded by the SRC-homology 1 (SH1) domain of the ABL component of BCR-ABL is undoubtedly the most crucial for oncogenic transformation. Other important motifs in the ABL portion are the protein-interaction SRC-homology 2 (SH2) and the C-terminal actin-binding domains. On the BCR moiety, the coiled-coil motif encoded by the first BCR exon is responsible for dimerization of the oncoprotein; a tyrosine at position 177 is crucial for the binding of adaptor proteins such as growth factor receptor�bound protein 2; and N-terminal phosphoserine and phosphothreonine residues are required for interaction with SH2-containing proteins, including ABL itself. Numerous substrates have been found to bind to BCR-ABL and to be tyrosine-phosphorylated by it (Table 2 and Figure 3), and the list of substrates keeps increasing. Associations with tyrosine phosphatases such as protein tyrosine phosphatase B1 (PTPB1) and Syp have also been reported, but these phosphatases may in fact limit the kinase activity of BCR-ABL, an effect that would oppose other BCR-ABL leukemogenic forces. However, most interactions and activation processes have been studied only in cell lines in vitro and under conditions of forced (ectopic) overexpression. In most cases, therefore, their very existence in primary leukemic cells and their relevance to the CML phenotype in vivo remain uncertain.
|
|
One of the most striking differences between the normal ABL protein and BCR-ABL is in their contrasting subcellular locations. The ABL protein is found in both the nucleus and the cytoplasm and can shuttle between these two compartments under the influence of its nuclear-localization signal and nuclear-export signal domains, whereas BCR-ABL is exclusively cytoplasmic. Nuclear ABL is an essentially proapoptotic protein, playing a key part in the cellular response to genotoxic stress. BCR-ABL, in contrast, is largely antiapoptotic and, although it retains the ABL nuclear-localization and nuclear-export sequences, seems unable to enter the nucleus. The main reason the BCR-ABL protein is retained in the cytoplasm is its constitutively activated tyrosine kinase.78 When the kinase is inhibited in vitro with imatinib and its nuclear export simultaneously blocked with leptomycin B, the oncoprotein enters the nucleus and is trapped there. Interestingly, when imatinib is removed and the now nuclear BCR-ABL is allowed to reactivate its tyrosine kinase, it is converted from an antiapoptotic to a proapoptotic protein and induces, rather than prevents, cell death. It has recently been shown that BCR-ABL also translocates to the nucleus in leukemia cells subjected to genotoxic stress and there slows DNA repair by interaction with effectors of the ataxia telangiectasia�related (ATR) protein, a phenomenon that may underlie the genomic instability of the CML clone.79
In the late 1980s, accumulating data on the mechanisms of BCR-ABL function set the scene for the design of molecularly targeted therapy. Since the tyrosine kinase is the effector part of the oncoprotein, it was obviously the most attractive target for inhibition. The aim was to design a small chemical compound that could compete with ATP for its binding site in the kinase domain. Whereas the normal binding of ATP allows BCR-ABL to phosphorylate selected tyrosine residues on its substrates, an ATP mimic occupying the binding pocket would not provide any phosphate group for transfer to the substrate. With its tyrosine residues in the unphosphorylated form, the substrate protein would not then undergo the required conformational change to allow it to associate with its downstream effector. The entire chain of downstream reactions would then be impeded, interrupting transmission of the oncogenic signal to the nucleus.
During the 1990s, a number of tyrosine kinase inhibitors were purified from natural substances (e.g., herbimycin A and genistein) or designed and synthesized in the laboratory (e.g., tyrphostins).80,81,82 However, by far the most successful inhibitor was imatinib, a small, 2-phenylaminopyrimidine molecule that, at micromolar concentrations, inhibits the kinase activity of all proteins that contain ABL, ABL-related gene (ARG) protein, or platelet-derived growth factor receptor, as well as the KIT receptor.83,84,85 This compound inhibits cellular growth and induces apoptosis in CML, both in vitro and in vivo.86,87,88
The importance of imatinib goes beyond its undoubted therapeutic value in CML. Because it was rationally designed to carry out a predetermined function and proved so successful, other candidate agents for targeted therapy in CML have been rapidly emerging; among them are adaphostin (an ABL tyrphostin), geldanamycin (an inhibitor of the BCR-ABL chaperone heat-shock protein 90), and the farnesyl transferase inhibitors (which prevent activation of RAS and other farnesylated oncoproteins). Each of these could prove clinically valuable if combined with imatinib, even after resistance to imatinib as a single agent has developed.89,90,91 Moreover, the combined use of imatinib and leptomycin B in a regimen similar to the experimental one developed by Vigneri and Wang78 may prove valuable for eliminating residual leukemia cells before autografting, a setting in which imatinib alone is not very effective.92 A family of SRC tyrosine kinase inhibitors (in particular, PD166326, PD173955, and PD180970) was also recently reported to be active against BCR-ABL and to inhibit the in vitro growth of primary CML progenitor cells.93
Because of the excitement they have generated, the synthetic signal-transduction inhibitors may, for the present, overshadow other potential antileukemia agents, but the development of drug resistance � already documented for imatinib94,95,96,97,98,99 � should be a reminder of the possible value of combining different molecular approaches. Although traditional oligodeoxynucleotide or ribozyme antisense molecules against BCR-ABL mRNA failed to fulfill their initial promise for the treatment of CML, better sequence design and chemical techniques have recently provided new ideas for the success of this approach, among them a dimeric ribozyme or "maxizyme"100 and RNA-interference strategies.101 Equally interesting is the possibility of expressing a single-chain antibody, or "intrabody," against ABL (or possibly BCR-ABL) by retroviral-mediated gene transfer into CML cells.102 Finally, attempts at immunization with molecularly engineered BCR-ABL fusion peptides or plasmid DNA should be considered a possible adjunct to debulking regimens with signal transduction inhibitors.
Graft-Versus-Leukemia Effect
The notion that the eradication of leukemia by allogeneic stem-cell transplantation does not depend entirely on the chemotherapy and radiotherapy given during the conditioning phase that precedes stem-cell infusion, but that it relies also on an ill-defined graft-versus-leukemia effect, has been generally accepted for some years.20,103 The concept that this graft-versus-leukemia effect may be mediated by allogeneic T cells derives support from four somewhat discrete lines of evidence � namely, the observations that the incidence of relapse in leukemia is inversely related to the incidence of GVHD, that relapse is more common after the transplantation of hematopoietic stem cells from syngeneic rather than allogeneic sibling donors, that depletion of T cells from the donor inoculum greatly increases the risk of relapse, and that the infusion of lymphocytes collected from the original donor can restore complete remission in a patient who has had a relapse after allografting, especially in the case of CML. These observations led logically to a search for the target antigen or antigens recognized by the allogeneic T cells.
Almost all the peptide sequences that make up the BCR-ABL protein are also present in the normal ABL or BCR proteins, but the junctional codons and their corresponding amino acids are unique and thus specific to leukemia. There is recent evidence that such leukemia-specific oligopeptides may be presented on the leukemia-cell surface in conjunction with HLA class II molecules.104 Alternatively, the target antigens for the graft-versus-leukemia effect could be lineage-specific rather than leukemia-specific. Cytotoxic T lymphocytes that recognize the PR-1 component of proteinase 3 or the Wilms' tumor 1 antigen, both of which are overexpressed in leukemia cells, kill CML cells in vitro but spare control cells from normal persons.105,106
Attempts are now being made to immunize patients against their own leukemia. A group at Memorial Sloan-Kettering Cancer Center, in New York, showed that patients with the e14a2 junction may generate cytotoxic T lymphocytes in response to immunization with an e14a2-derived junctional oligopeptide presented in conjunction with HLA molecules and a suitable adjuvant agent.107 Cytotoxic T lymphocytes directed against known antigens could prove valuable in treating minimal residual disease but may not be so effective in dealing with large quantities of leukemia.
Treatment Options
Alternatives to Transplantation
The options for treating patients with newly diagnosed CML have changed fundamentally since the introduction of imatinib. Until recently, interferon alfa was generally accepted as the best single agent for treating CML in the chronic phase in patients who were not eligible for allogeneic stem-cell transplantation. The drug may cause a wide range of side effects, especially in older persons, but it induces complete or nearly complete cytogenetic responses in 10 to 30 percent of patients and probably prolongs survival to a greater extent than hydroxyurea.108 A French multicenter study showed that survival among patients treated with the combination of interferon alfa and cytarabine was superior to that among patients treated with interferon alfa alone,109 but that potentially important observation was not confirmed in a more recent study.110
Imatinib was first used in 1998 to treat patients who were judged to have disease refractory to treatment with interferon alfa or who could not tolerate interferon alfa.84 Imatinib induced complete hematologic responses in more than 95 percent of patients with CML resistant to interferon alfa but still in the chronic phase, and it induced major cytogenetic responses in 40 to 50 percent.17 Preliminary analyses of the results of treatment in relatively large numbers of patients treated in six countries have shown that event-free survival and overall survival are better than might be expected with alternative therapies.111 One recent study suggests that patients who achieve good cytogenetic responses live longer than historical control patients.112 Among patients treated in the accelerated phase of the disease, overall survival and event-free survival appear to be superior to those in historical controls.113 In contrast, the majority of patients with disease in blastic transformation have a response to imatinib, but these responses are short-lived, and no definite survival benefit can be discerned.114,115 The drug can cause nausea, headache, rashes, fluid retention, clinically significant cytopenia, and other side effects, but these problems are generally manageable and seem to be appreciably less troublesome than those associated with interferon alfa.84
Although data on cytogenetic responses as a surrogate marker of survival suggest that imatinib prolongs the survival of patients treated in the chronic phase of disease, this conclusion must for the present be regarded as unproved. For this reason, the Food and Drug Administration recommended to the manufacturers of the drug that they undertake a multicenter study in which imatinib is compared prospectively with the combination of interferon alfa and cytarabine in previously untreated patients with CML. The study was undertaken, and patients were recruited in 2001. According to a recent interim assessment of the results, the rate of complete cytogenetic response in the imatinib group is 74 percent,18 and some of those patients have achieved a molecular remission (the absence of detectable BCR-ABL transcripts in the blood). As a consequence, the study has been complicated by the desire of some patients in the interferon alfa�cytarabine group to cross over to the imatinib group. It is hoped, nonetheless, that the trial will show before too long whether treatment with imatinib prolongs the survival of patients with newly diagnosed CML.
Although resistance to imatinib as a single agent seems to be rare in patients treated in the chronic phase of disease, resistance does eventually develop in the majority of patients treated in the advanced phase. The mechanism of this acquired resistance is of great interest. Studies in vitro of resistant cell lines have shown amplification of the BCR-ABL gene in association with overexpression of the oncoprotein.94 Moreover, in the clinic, a proportion of patients whose disease has become resistant to imatinib have acquired point mutations in the ABL kinase domain that lead to specific amino acid substitutions that could, theoretically, interfere with the binding of imatinib in the kinase pocket97 98,116,117,118,119,120,121,122 (Table 3). Presumably, these mutations do not prevent the kinase from accepting ATP and thus phosphorylating the substrates that generate the CML phenotype. In a substantial proportion of patients, however, resistance to imatinib is probably not due to BCR-ABL overexpression or mutations in the ABL kinase domain, particularly because some of the point mutations detected do not result in loss of sensitivity to imatinib.123 In these cases, the resistant phenotype is probably due to the capacity of CML cells to recruit signal-transduction pathways that bypass the block or, as recently demonstrated by in vitro mutagenesis, to point mutations outside the kinase domain.124
|
Stem-Cell Transplantation
It is now generally accepted that allogeneic hematopoietic stem-cell transplantation can cure CML in selected patients,19,125,126,127 but stem-cell transplantation19,127 is still associated with an appreciable risk of complications and death, due principally to GVHD and opportunistic infections. The decision about whether to offer transplantation to a given patient is aided to some degree by knowledge that the factors that influence the risk of transplantation-related death are now reasonably well defined and include the patient's age, the phase of disease, the duration of disease, the degree of donor�recipient histocompatibility, and the donor's sex.128,129 Thus, for example, a young patient with newly diagnosed CML in the chronic phase who has an HLA-identical sibling donor can expect to fare much better after transplantation than an older patient with accelerated-phase disease who has a less well matched, unrelated donor.
The recognition that the graft-versus-leukemia effect20 plays a major part in the eradication of CML after allografting led to the concept that the toxic effects associated with allografting could be substantially reduced by lowering the overall intensity of the pretransplantation conditioning regimen. This approach involves substantial reductions in the dose of cytotoxic drugs or radiation used in conditioning, with emphasis instead on the use of immunosuppressive agents. At the same time, the number of hematopoietic stem cells and lymphocytes transfused to the patient is maximized to ensure engraftment and an optimal graft-versus-leukemia effect. A wide variety of regimens have been devised for such transplantations, which have been called "nonmyeloablative," "reduced-intensity conditioning transplantation," and "mini-transplantation."130,131,132 By reducing the intensity of conditioning in this way, it may be possible to offer transplantation to patients who would not otherwise be eligible because of older age or the presence of concomitant disease. Some patients who have received this type of treatment have had durable cytogenetic and molecular remissions,132 but long-term results are still difficult to assess.
Strategies for Decision Making
The questions of whether one should recommend allogeneic stem-cell transplantation to patients with CML in the chronic phase and, if so, to which patients remain challenging.133 One possible approach is to balance the perceived benefits and risks of transplantation against the likelihood of long-term survival if the best available nontransplantation therapy is used. The results of nontransplantation therapy in a given patient may be predicted in very general terms by reference to scoring systems devised by Sokal and colleagues134 and subsequently updated by Hasford and colleagues.135 Because neither the results of transplantation nor the long-term outcome of treatment with imatinib or imatinib-containing combinations can be accurately predicted, one possibility would be to offer patients with newly diagnosed CML a trial of treatment with imatinib and then to offer transplantation to those in whom a complete or nearly complete cytogenetic response has not been achieved by six to nine months, provided that the estimated risk of transplantation-related death is reasonably low (e.g., less than 20 percent); other patients could be offered alternative nontransplantation therapy.133 The best approach to managing CML in young patients who have a suitable transplant donor may be clearer in one or two years, when we have gained more experience in the use of imatinib.
Future Prospects
Although much has been achieved, many important issues pertaining to the biology and treatment of CML remain unresolved. To mention just a few, we know little of the mechanisms that cause the chromosomal rearrangement. We still need to clarify how deregulation of signal transduction by the BCR-ABL oncoprotein leads to the proliferative advantage of the Ph-positive clone. We need a much better understanding of the molecular basis of disease progression. Of tremendous value would be certain identification of the target antigens in the graft-versus-leukemia effect. In therapeutic terms, we need to define as rapidly as possible the true clinical potential of imatinib and to ascertain whether combining this agent with other signal-transduction inhibitors, other cytotoxic drugs, or differentiating agents can improve its efficacy. We need to know whether immunizing patients with CML can prolong their survival or contribute to the eradication of disease. It seems that at least some of these problems will be solved within the next five years.
We are indebted to our clinical and nonclinical colleagues
who contributed many of the ideas and the bulk of the data included
in this review, especially colleagues at Hammersmith Hospital, including
Professors Jane Apperley and Myrtle Gordon and Drs. David Barnes,
Francesco Dazzi, Edward Kanfer, Eduardo Olavarria, David Marin, Amin
Rahemtulla, and Alex Tipping.
Source Information
From the Department of Haematology, Faculty of Medicine, Imperial College London, London.
Address reprint requests to Dr. Goldman at Hammersmith Hospital, Imperial College London, Du Cane Rd., London W12 0NN, United Kingdom, or at [email protected].
References
- Geary CG. The story of chronic myeloid leukaemia. Br J Haematol 2000;110:2-11. [CrossRef][ISI][Medline]
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960;132:1497-1497. [ISI]
- Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973;243:290-293. [ISI][Medline]
- Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985;315:550-554. [ISI][Medline]
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med
1999;340:1330-1340.
[Full Text] - Verfaillie CM. Chronic myelogenous leukemia: from pathogenesis to therapy. J Hematother 1999;8:3-13. [CrossRef][ISI][Medline]
- Faderl S, Talpaz M, Estrov Z, O'Brien S, Kurzrock R, Kantarjian
HM. The biology of chronic myeloid leukemia. N Engl J Med 1999;341:164-172.
[Full Text] - Deininger MW, Goldman JM, Melo JV. The molecular biology of
chronic myeloid leukemia. Blood 2000;96:3343-3356.
[Full Text] - Eaves CJ, Eaves AC. Stem cell kinetics. Baillieres Clin Haematol 1997;10:233-257. [Medline]
- Eaves CJ, Eaves AC. Progenitor cell dynamics. In: Carella AM, Daley GQ, Eaves CJ, Goldman JM, Hehlmann R, eds. Chronic myeloid leukaemia: biology and treatment. London: Martin Dunitz, 2001:73-100.
- Jiang X, Lopez A, Holyoake T, Eaves A, Eaves C. Autocrine
production and action of IL-3 and granulocyte colony-stimulating factor in
chronic myeloid leukemia. Proc Natl Acad Sci U S A 1999;96:12804-12809.
[Abstract/Full Text] - Holyoake TL, Jiang X, Jorgensen HG, et al. Primitive quiescent
leukemic cells from patients with chronic myeloid leukemia spontaneously
initiate factor-independent growth in vitro in association with
up-regulation of expression of interleukin-3. Blood 2001;97:720-728.
[Abstract/Full Text] - Bedi A, Zehnbauer BA, Barber JP, Sharkis SJ, Jones RJ.
Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood
1994;83:2038-2044.
[Abstract/Full Text] - Graham SM, Jorgensen HG, Allan E, et al. Primitive, quiescent,
Philadelphia-positive stem cells from patients with chronic myeloid leukemia
are insensitive to STI571 in vitro. Blood 2002;99:319-325.
[Abstract/Full Text] - Coulombel L, Kalousek DK, Eaves CJ, Gupta CM, Eaves AC. Long-term marrow culture reveals chromosomally normal hematopoietic progenitor cells in patients with Philadelphia chromosome-positive chronic myelogenous leukemia. N Engl J Med 1983;308:1493-1498. [Abstract]
- Carella AM, Podesta M, Frassoni F, et al. Collection of `normal' blood repopulating cells during early hemopoietic recovery after intensive conventional chemotherapy in chronic myelogenous leukemia. Bone Marrow Transplant 1993;12:267-271. [ISI][Medline]
- Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a
specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid
leukemia. N Engl J Med 2001;344:1031-1037.
[Abstract/Full Text] - O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with
interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic
myeloid leukemia. N Engl J Med 2003;348:994-1004.
[Abstract/Full Text] - van Rhee F, Szydlo RM, Hermans J, et al. Long-term results after allogeneic bone marrow transplantation for chronic myelogenous leukemia in chronic phase: a report from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant 1997;20:553-560. [CrossRef][ISI][Medline]
- Barrett AJ, Malkovska V. Graft-versus-leukaemia: understanding and using the alloimmune response to treat haematological malignancies. Br J Haematol 1996;93:754-761. [ISI][Medline]
- Cross NCP. Assessing residual disease. In: Goldman JM, ed. Baillieres Clin Haematol 1997;10:389-403. [Medline]
- Chomel JC, Brizard F, Veinstein A, et al. Persistence of
BCR-ABL genomic rearrangement in chronic myeloid leukemia patients in
complete and sustained cytogenetic remission after interferon-alpha therapy
or allogeneic bone marrow transplantation. Blood 2000;95:404-408.
[Abstract/Full Text] - Chase A, Parker S, Kaeda J, Sivalingam R, Cross NC, Goldman JM.
Absence of host-derived cells in the blood of patients in remission after
allografting for chronic myeloid leukemia. Blood 2000;96:777-778.
[Full Text] - Deininger M, Lehmann T, Krahl R, Hennig E, Muller C, Niederwieser D. No evidence for persistence of BCR-ABL-positive cells in patients in molecular remission after conventional allogenic transplantation for chronic myeloid leukemia. Blood 2000;96:779-780. [ISI][Medline]
- Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of
major bcr-abl gene expression at a very low level in blood cells of some
healthy individuals. Blood 1995;86:3118-3122.
[Abstract/Full Text] - Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The
presence of typical and atypical BCR-ABL fusion genes in leukocytes of
normal individuals: biologic significance and implications for the
assessment of minimal residual disease. Blood 1998;92:3362-3367.
[Abstract/Full Text] - Preston DL, Kusumi S, Tomonaga M, et al. Cancer incidence in atomic bomb survivors. III. Leukemia, lymphoma and multiple myeloma, 1950-1987. Radiat Res 1994;137:Suppl:S68-S97. [Erratum, Radiat Res 1994;139:129.] [ISI][Medline]
- Deininger MW, Bose S, Gora-Tybor J, Yan XH, Goldman JM, Melo JV. Selective induction of leukemia-associated fusion genes by high-dose ionizing radiation. Cancer Res 1998;58:421-425. [Abstract]
- Neves H, Ramos C, da Silva MG, Parreira A, Parreira L. The
nuclear topography of ABL, BCR, PML, and RARalpha genes: evidence for gene
proximity in specific phases of the cell cycle and stages of hematopoietic
differentiation. Blood 1999;93:1197-1207.
[Abstract/Full Text] - Saglio G, Storlazzi CT, Giugliano E, et al. A 76-kb duplicon
maps close to the BCR gene on chromosome 22 and the ABL gene on chromosome
9: possible involvement in the genesis of the Philadelphia chromosome
translocation. Proc Natl Acad Sci U S A 2002;99:9882-9887.
[Abstract/Full Text] - Melo JV, Gordon DE, Cross NC, Goldman JM. The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood 1993;81:158-165. [Abstract]
- Sinclair PB, Nacheva EP, Leversha M, et al. Large deletions at
the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup
of patients with chronic myeloid leukemia. Blood 2000;95:738-743.
[Abstract/Full Text] - Huntly BJ, Reid AG, Bench AJ, et al. Deletions of the
derivative chromosome 9 occur at the time of the Philadelphia translocation
and provide a powerful and independent prognostic indicator in chronic
myeloid leukemia. Blood 2001;98:1732-1738.
[Abstract/Full Text] - de la Fuente J, Merx K, Steer J, et al. ABL-BCR expression does
not correlate with deletions on the derivative chromosome 9 or survival in
chronic myeloid leukemia. Blood 2001;98:2879-2880.
[Full Text] - Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol 2002;107:76-94. [CrossRef][ISI][Medline]
- Feinstein E, Cimino G, Gale RP, et al. p53 In chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci U S A 1991;88:6293-6297. [Abstract]
- Sill H, Goldman JM, Cross NC. Homozygous deletions of the p16
tumor-suppressor gene are associated with lymphoid transformation of chronic
myeloid leukemia. Blood 1995;85:2013-2016.
[Abstract/Full Text] - Towatari M, Adachi K, Kato H, Saito H. Absence of the human retinoblastoma gene product in the megakaryoblastic crisis of chronic myelogenous leukemia. Blood 1991;78:2178-2181. [Abstract]
- Ogawa S, Mitani K, Kurokawa M, et al. Abnormal expression of Evi-1 gene in human leukemias. Hum Cell 1996;9:323-332. [Medline]
- Cortes JE, Talpaz M, Beran M, et al. Philadelphia chromosome-negative chronic myelogenous leukemia with rearrangement of the breakpoint cluster region: long-term follow-up results. Cancer 1995;75:464-470. [ISI][Medline]
- Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 1994;77:307-316. [ISI][Medline]
- Reiter A, Sohal J, Kulkarni S, et al. Consistent fusion of
ZNF198 to the fibroblast growth factor receptor-1 in the t(8;13) (p11;q12)
myeloproliferative syndrome. Blood 1998;92:1735-1742.
[Abstract/Full Text] - Wilbanks AM, Mahajan S, Frank DA, Druker BJ, Gilliland DG, Carroll M. TEL/PDGFbetaR fusion protein activates STAT1 and STAT5: a common mechanism for transformation by tyrosine kinase fusion proteins. Exp Hematol 2000;28:584-593. [CrossRef][ISI][Medline]
- Smedley D, Demiroglu A, Abdul-Rauf M, et al. ZNF198-FGFR1 transforms Ba/F3 cells to growth factor independence and results in high level tyrosine phosphorylation of STATS 1 and 5. Neoplasia 1999;1:349-355. [CrossRef][Medline]
- Steer EJ, Cross NC. Myeloproliferative disorders with translocations of chromosome 5q31-35: role of the platelet-derived growth factor receptor beta. Acta Haematol 2002;107:113-122. [CrossRef][ISI][Medline]
- Fialkow PJ, Martin PJ, Najfeld V, Penfold GK, Jacobson RJ, Hansen JA. Evidence for a multistep pathogenesis of chronic myelogenous leukemia. Blood 1981;58:158-163. [Abstract]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990;247:824-830. [ISI][Medline]
- Pear WS, Miller JP, Xu L, et al. Efficient and rapid induction
of a chronic myelogenous leukemia-like myeloproliferative disease in mice
receiving P210 bcr/abl-transduced bone marrow. Blood 1998;92:3780-3792.
[Abstract/Full Text] - Zhang X, Ren R. Bcr-Abl efficiently induces a
myeloproliferative disease and production of excess interleukin-3 and
granulocyte-macrophage colony-stimulating factor in mice: a novel model for
chronic myelogenous leukemia. Blood 1998;92:3829-3840.
[Abstract/Full Text] - Pane F, Frigeri F, Sindona M, et al. Neutrophilic-chronic
myeloid leukemia: a distinct disease with a specific molecular marker (BCR/ABL
with C3/A2 junction). Blood 1996;88:2410-2414. [Erratum, Blood
1997;89:4244.]
[Abstract/Full Text] - Melo JV, Myint H, Galton DA, Goldman JM. P190BCR-ABL chronic myeloid leukaemia: the missing link with chronic myelomonocytic leukaemia? Leukemia 1994;8:208-211. [ISI][Medline]
- Melo JV. The diversity of BCR-ABL fusion proteins and their
relationship to leukemia phenotype. Blood 1996;88:2375-2384.
[Full Text] - Pluk H, Dorey K, Superti-Furga G. Autoinhibition of c-Abl. Cell 2002;108:247-259. [ISI][Medline]
- Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JY.
Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear
transport. Proc Natl Acad Sci U S A 1996;93:15174-15179.
[Abstract/Full Text] - Salgia R, Li JL, Ewaniuk DS, et al. BCR/ABL induces multiple
abnormalities of cytoskeletal function. J Clin Invest 1997;100:46-57.
[Abstract/Full Text] - McWhirter JR, Galasso DL, Wang JY. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol 1993;13:7587-7595. [Abstract]
- Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell 2003;12:27-37. [ISI][Medline]
- Barnes DJ, Melo JV. Cytogenetic and molecular genetic aspects of chronic myeloid leukaemia. Acta Haematol 2002;108:180-202. [CrossRef][ISI][Medline]
- Li S, Couvillon AD, Brasher BB, Van Etten RA. Tyrosine
phosphorylation of Grb2 by Bcr/Abl and epidermal growth factor receptor: a
novel regulatory mechanism for tyrosine kinase signaling. EMBO J
2001;20:6793-6804.
[Abstract/Full Text] - Yamanashi Y, Baltimore D. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell 1997;88:205-211. [ISI][Medline]
- Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ.
Crkl is the major tyrosine-phosphorylated protein in neutrophils from
patients with chronic myelogenous leukemia. J Biol Chem
1994;269:22925-22928.
[Abstract/Full Text] - Sattler M, Salgia R, Okuda K, et al. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3' kinase pathway. Oncogene 1996;12:839-846. [ISI][Medline]
- Tauchi T, Boswell HS, Leibowitz D, Broxmeyer HE. Coupling between p210bcr-abl and Shc and Grb2 adaptor proteins in hematopoietic cells permits growth factor receptor-independent link to ras activation pathway. J Exp Med 1994;179:167-175. [Abstract]
- Salgia R, Brunkhorst B, Pisick E, et al. Increased tyrosine phosphorylation of focal adhesion proteins in myeloid cell lines expressing p210BCR/ABL. Oncogene 1995;11:1149-1155. [ISI][Medline]
- Salgia R, Li JL, Lo SH, et al. Molecular cloning of human
paxillin, a focal adhesion protein phosphorylated by P210BCR/ABL. J Biol
Chem 1995;270:5039-5047.
[Abstract/Full Text] - Gotoh A, Miyazawa K, Ohyashiki K, et al. Tyrosine phosphorylation and activation of focal adhesion kinase (p125FAK) by BCR-ABL oncoprotein. Exp Hematol 1995;23:1153-1159. [ISI][Medline]
- Ernst TJ, Slattery KE, Griffin JD. p210Bcr/Abl and p160v-Abl
induce an increase in the tyrosine phosphorylation of p93c-Fes. J Biol Chem
1994;269:5764-5769.
[Abstract/Full Text] - Gotoh A, Miyazawa K, Ohyashiki K, Toyama K. Potential molecules implicated in downstream signaling pathways of p185BCR-ABL in Ph+ ALL involve GTPase-activating protein, phospholipase C-gamma 1, and phosphatidylinositol 3'-kinase. Leukemia 1994;8:115-120. [ISI][Medline]
- Shi CS, Tuscano JM, Witte ON, Kehrl JH. GCKR links the Bcr-Abl
oncogene and Ras to the stress-activated protein kinase pathway. Blood
1999;93:1338-1345.
[Abstract/Full Text] - Skorski T, Kanakaraj P, Nieborowska-Skorska M, et al.
Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is
required for the growth of Philadelphia chromosome-positive cells. Blood
1995;86:726-736.
[Abstract/Full Text] - Dai Z, Quackenbush RC, Courtney KD, et al. Oncogenic Abl and
Src tyrosine kinases elicit the ubiquitin-dependent degradation of target
proteins through a Ras-independent pathway. Genes Dev 1998;12:1415-1424.
[Abstract/Full Text] - Tauchi T, Feng GS, Shen R, et al. SH2-containing
phosphotyrosine phosphatase Syp is a target of p210bcr-abl tyrosine kinase.
J Biol Chem 1994;269:15381-15387.
[Abstract/Full Text] - Wisniewski D, Strife A, Swendeman S, et al. A novel
SH2-containing phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase
(SHIP2) is constitutively tyrosine phosphorylated and associated with src
homologous and collagen gene (SHC) in chronic myelogenous leukemia
progenitor cells. Blood 1999;93:2707-2720.
[Abstract/Full Text] - Reuther GW, Fu H, Cripe LD, Collier RJ, Pendergast AM. Association of the protein kinases c-Bcr and Bcr-Abl with proteins of the 14-3-3 family. Science 1994;266:129-133. [ISI][Medline]
- Bhat A, Kolibaba K, Oda T, Ohno-Jones S, Heaney C, Druker BJ.
Interactions of CBL with BCR-ABL and CRKL in BCR-ABL-transformed myeloid
cells. J Biol Chem 1997;272:16170-16175.
[Abstract/Full Text] - Matsuguchi T, Inhorn RC, Carlesso N, Xu G, Druker B, Griffin JD. Tyrosine phosphorylation of p95Vav in myeloid cells is regulated by GM-CSF, IL-3, and steel factor and is constitutively increased by p210BCR/ABL. EMBO J 1995;14:257-265. [Abstract]
- Dai Z, Kerzic P, Schroeder WG, McNiece IK. Deletion of the Src
homology 3 domain and C-terminal proline-rich sequences in Bcr-Abl prevents
Abl interactor 2 degradation and spontaneous cell migration and impairs
leukemogenesis. J Biol Chem 2001;276:28954-28960.
[Abstract/Full Text] - Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med 2001;7:228-234. [CrossRef][ISI][Medline]
- Dierov JK, Dierova R, Carroll M. BCR/ABL translocates to the nucleus after DNA damage and interacts with the ataxia-telangiectasia mutant (ATM) protein to modify DNA repair. Blood 2002;100:204a-204a. abstract.
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science 1995;267:1782-1788. [ISI][Medline]
- Druker BJ, Lydon NB. Lessons learned from the development of an
Abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin
Invest 2000;105:3-7.
[Full Text] - Traxler P, Bold G, Buchdunger E, et al. Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev 2001;21:499-512. [CrossRef][ISI][Medline]
- Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 1996;56:100-104. [Abstract]
- Savage DG, Antman KH. Imatinib mesylate -- a new oral targeted
therapy. N Engl J Med 2002;346:683-693.
[Full Text] - Okuda K, Weisberg E, Gilliland DG, Griffin JD. ARG tyrosine
kinase activity is inhibited by STI571. Blood 2001;97:2440-2448.
[Abstract/Full Text] - Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561-566. [ISI][Medline]
- Deininger MWN, Goldman JM, Lydon NB, Melo JV. The tyrosine
kinase inhibitor CGP57148B selectively inhibits the growth of
BCR-ABL-positive cells. Blood 1997;90:3691-3698.
[Abstract/Full Text] - le Coutre P, Mologni L, Cleris L, et al. In vivo eradication of
human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J Natl
Cancer Inst 1999;91:163-168.
[Abstract/Full Text] - Mow BM, Chandra J, Svingen PA, et al. Effects of the Bcr/abl
kinase inhibitors STI571 and adaphostin (NSC 680410) on chronic myelogenous
leukemia cells in vitro. Blood 2002;99:664-671.
[Abstract/Full Text] - Gorre ME, Ellwood-Yen K, Chiosis G, Rosen N, Sawyers CL.
BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant
chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL
chaperone heat shock protein 90. Blood 2002;100:3041-3044.
[Abstract/Full Text] - Hoover RR, Mahon FX, Melo JV, Daley GQ. Overcoming STI571
resistance with the farnesyl transferase inhibitor SCH66336. Blood
2002;100:1068-1071.
[Abstract/Full Text] - Schultheis B, Yong ASM, Goldman JM, Melo JV. The combined use of STI571 and leptomycin-B (LMB) for in vitro purging of leukemic cells in autografts for CML. Blood 2001;98:618a-618a. abstract. [CrossRef]
- Wisniewski D, Lambek CL, Liu C, et al. Characterization of
potent inhibitors of the Bcr-Abl and the c-kit receptor tyrosine kinases.
Cancer Res 2002;62:4244-4255.
[Abstract/Full Text] - Mahon FX, Deininger MW, Schultheis B, et al. Selection and
characterization of BCR-ABL positive cell lines with differential
sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of
resistance. Blood 2000;96:1070-1079.
[Abstract/Full Text] - le Coutre P, Tassi E, Varella-Garcia M, et al. Induction of
resistance to the Abelson inhibitor STI571 in human leukemic cells through
gene amplification. Blood 2000;95:1758-1756.
[Abstract/Full Text] - Weisberg E, Griffin JD. Mechanism of resistance to the ABL
tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell
lines. Blood 2000;95:3498-3505.
[Abstract/Full Text] - Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to
STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification.
Science 2001;293:876-880.
[Abstract/Full Text] - Barthe C, Cony-Makhoul P, Melo JV, Mahon JR. Roots of clinical resistance to STI-571 cancer therapy. Science 2001;293:2163-2163. [CrossRef][Medline]
- Hochhaus A, Kreil S, Corbin A, et al. Roots of clinical resistance to STI-571 cancer therapy. Science 2001;293:2163-2163. [CrossRef][Medline]
- Tanabe T, Kuwabara T, Warashina M, Tani K, Taira K, Asano S. Oncogene inactivation in a mouse model. Nature 2000;406:473-474. [CrossRef][ISI][Medline]
- Wilda M, Fuchs U, Wossmann W, Borkhardt A. Killing of leukemic cells with a BCR/ABL fusion gene by RNA interference (RNAi). Oncogene 2002;21:5716-5724. [CrossRef][ISI][Medline]
- Xu D, Song J, Li D, Verfaillie CM, Zhao RCH. Anti-ABL tyrosine kinase intrabody promotes apoptosis in K562 cells. Blood 2001;98:146a-146a. abstract. [CrossRef]
- Barrett AJ. Allogeneic stem cell transplantation for chronic myeloid leukemia. Semin Hematol 2003;40:59-71. [CrossRef][ISI][Medline]
- Clark RE, Dodi IA, Hill SC, et al. Direct evidence that
leukemic cells present HLA-associated immunogenic peptides derived from the
BCR-ABL b3a2 fusion protein. Blood 2001;98:2887-2893.
[Abstract/Full Text] - Molldrem JJ, Lee PP, Wang C, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med 2000;6:1018-1023. [CrossRef][ISI][Medline]
- Gao L, Bellantuono I, Elsasser A, et al. Selective elimination
of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for
WT1. Blood 2000;95:2198-2203.
[Abstract/Full Text] - Pinilla-Ibarz J, Cathcart K, Korontsvit T, et al. Vaccination
of patients with chronic myelogenous leukemia with bcr-abl oncogene
breakpoint fusion peptides generates specific immune responses. Blood
2000;95:1781-1787.
[Abstract/Full Text] - Chronic Myeloid Leukemia Trialists' Collaborative Group.
Interferon alfa versus chemotherapy for chronic myeloid leukemia: a
meta-analysis of seven randomized trials. J Natl Cancer Inst
1997;89:1616-1620.
[Abstract/Full Text] - Guilhot F, Chastang C, Michallet M, et al. Interferon alfa-2b
combined with cytarabine versus interferon alone in chronic myelogenous
leukemia. N Engl J Med 1997;337:223-229.
[Abstract/Full Text] - Baccarani M, Rosti G, de Vivo A, et al. A randomized study of
interferon-alpha versus interferon-alpha and low-dose arabinosyl cytosine in
chronic myeloid leukemia. Blood 2002;99:1527-1535.
[Abstract/Full Text] - Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and
cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia.
N Engl J Med 2002;346:645-652. [Erratum, N Engl J Med 2002;346:1923.]
[Abstract/Full Text] - Marin D, Marktel S, Szydlo R, et al. Survival of patients with chronic-phase chronic myeloid leukaemia on imatinib after failure on interferon alfa. Lancet 2003;362:617-619. [CrossRef][ISI][Medline]
- Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces
durable hematologic and cytogenetic responses in patients with accelerated
phase chronic myeloid leukemia: results of a phase 2 study. Blood
2002;99:1928-1937.
[Abstract/Full Text] - Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a
specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of
chronic myeloid leukemia and acute lymphoblastic leukemia with the
Philadelphia chromosome. N Engl J Med 2001;344:1038-1042. [Erratum, N Engl J
Med 2001;345:232.]
[Abstract/Full Text] - Wadhwa J, Szydlo RM, Apperley JF, et al. Factors affecting
duration of survival after onset of blastic transformation of chronic
myeloid leukemia. Blood 2002;99:2304-2309.
[Abstract/Full Text] - von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet 2002;359:487-491. [CrossRef][ISI][Medline]
- Branford S, Rudzki Z, Walsh S, et al. High frequency of point
mutations clustered within the adenosine triphosphate-binding region of BCR/ABL
in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic
leukemia who develop imatinib (STI571) resistance. Blood 2002;99:3472-3475.
[Abstract/Full Text] - Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al.
Several types of mutations of the Abl gene can be found in chronic myeloid
leukemia patients resistant to STI571, and they can pre-exist to the onset
of treatment. Blood 2002;100:1014-1018.
[Abstract/Full Text] - Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002;2:117-125. [CrossRef][ISI][Medline]
- Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002;16:2190-2196. [CrossRef][ISI][Medline]
- Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL
mutations in patients with CML treated with imatinib is virtually always
accompanied by clinical resistance, and mutations in the ATP
phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood
2003;102:276-283.
[Abstract/Full Text] - Hofmann WK, Jones LC, Lemp NA, et al. Ph(+) acute
lymphoblastic leukemia resistant to the tyrosine kinase inhibitor STI571 has
a unique BCR-ABL gene mutation. Blood 2002;99:1860-1862.
[Abstract/Full Text] - Corbin AS, La Rosee P, Stoffregen EP, Druker BJ, Deininger MW.
Several Bcr-Abl kinase domain mutants associated with imatinib mesylate
resistance remain sensitive to imatinib. Blood 2003;101:4611-4614.
[Abstract/Full Text] - Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003;112:831-843. [ISI][Medline]
- Thomas ED, Clift RA, Fefer A, et al. Marrow transplantation for the treatment of chronic myelogenous leukemia. Ann Intern Med 1986;104:155-163. [ISI][Medline]
- Goldman JM, Szydlo R, Horowitz MM, et al. Choice of pretransplant treatment and timing of transplants for chronic myelogenous leukemia in chronic phase. Blood 1993;82:2235-2238. [Abstract]
- Gale RP, Hehlmann R, Zhang MJ, et al. Survival with bone
marrow transplantation versus hydroxyurea or interferon for chronic
myelogenous leukemia. Blood 1998;91:1810-1819.
[Abstract/Full Text] - Gratwohl A, Hermans J, Goldman JM, et al. Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Lancet 1998;352:1087-1092. [CrossRef][ISI][Medline]
- Passweg JR, Walker I, Sobocinski KA, et al. Validation of the EBMT risk score for recipients of allogeneic stem cell transplants for CML. Bone Marrow Transplant 2002;27:Suppl 2:S33-S33. abstract.
- Giralt S, Thall PF, Khouri I, et al. Melphalan and purine
analog-containing preparative regimens: reduced-intensity conditioning for
patients with hematologic malignancies undergoing allogeneic progenitor cell
transplantation. Blood 2001;97:631-637.
[Abstract/Full Text] - Or R, Shapira MY, Resnick I, et al. Nonmyeloablative
allogeneic stem cell transplantation for the treatment of chronic myeloid
leukemia in first chronic phase. Blood 2003;101:441-445.
[Abstract/Full Text] - Uzunel M, Mattsson J, Brune M, Johansson JE, Aschan J, Ringden
O. Kinetics of minimal residual disease and chimerism in patients with
chronic myeloid leukemia after nonmyeloablative conditioning and allogeneic
stem cell transplantation. Blood 2003;101:469-472.
[Abstract/Full Text] - Goldman JM, Druker BJ. Chronic myeloid leukemia: current
treatment options. Blood 2001;98:2039-2042.
[Abstract/Full Text] - Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in "good-risk" chronic granulocytic leukemia. Blood 1984;63:789-799. [Abstract]
- Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic
score for survival of patients wtih chronic myeloid leukemia treated with
interferon alfa. J Natl Cancer Inst 1998;90:850-858.
[Abstract/Full Text]