Editorial
NEJM
| Volume 348:845-847 | February 27, 2003 | Number 9 |
|---|
Giancarlo Marra, M.D., Ph.D., and Josef Jiricny, Ph.D.
In this issue of the Journal, Sieber et al.1 report that up to 30 percent of cases of multiple colorectal adenomas (15 to 100 adenomas) might be linked to biallelic mutations in the MYH gene. What are the implications of this finding for the diagnosis and management of this disease?
Isolated adenomatous polyps are found in the colon in many persons, especially after 50 years of age. Since 5 to 10 percent of these lesions undergo malignant transformation over the course of several years, the endoscopist will reduce the risk of cancer by removing them. In the rare instances in which the colon contains multiple synchronous adenomas, the endoscopist's first reaction will be not to excise the lesions, but to count them. This is more than an arithmetic exercise: the number of polyps can have a key role in guiding the management of the disease.
Studies in humans and in mouse models have shown that the course of the disease can be directly related to the number of adenomas, because different ranges are frequently linked to specific genotypes. Thus, 50 to 80 percent of cases of adenomatous polyposis � a condition in which the patient presents with 100 to 1000 adenomas (and, in severe cases, even more) � can be classified as familial adenomatous polyposis, an autosomal dominant trait linked with germ-line mutations in the adenomatous polyposis coli gene (APC). APC encodes a tumor-suppressor protein, a key member of the Wnt signaling pathway, that controls proliferation of the colonic epithelial cells. APC mutations in families with familial adenomatous polyposis are clustered in several regions; most of these mutations result in inactivating truncations of the APC polypeptide. In contrast to patients with adenomatous polyposis, patients with multiple colorectal adenomas have fewer than 100 adenomas and are older at the onset of disease. Five to 10 percent of these cases, classified as attenuated adenomatous polyposis coli, are also linked to germ-line mutations in the APC gene, although these mutations tend to lie outside of the regions of mutational clusters.2,3,4 The position of the APC mutation appears to dictate not only the severity of the disease, but also the incidence of certain extracolonic manifestations, such as congenital hypertrophy of the retinal pigment epithelium.5 But what of the remaining cases of adenomatous polyposis and multiple colorectal adenomas that are not associated with APC mutations?
Recent evidence suggests that a substantial proportion of cases of
multiple colorectal adenomas, perhaps as many as 30 percent of those
with 15 to 100 polyps, might be associated with a novel type of
DNA-repair defect. The first hint as to its nature came from an
analysis of somatic APC mutations in the polyps from siblings
in a family with multiple adenomas6 and in those
from seven unrelated patients with polyposis.7
In both of these studies, the mutations identified in the adenomas
were transversions of a guanine�cytosine pair to a thymine�adenine
pair (G:C![]() T:A), unlike the
germ-line mutations in familial adenomatous polyposis and attenuated
adenomatous polyposis coli. Since this type of transversion
represents a known "footprint" of oxidative DNA damage, the
authors decided to study three genes encoding polypeptides that are
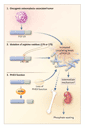
necessary for efficient processing of this type of damage (Figure
1). The first, MTH, encodes a polypeptide that
"sanitizes" oxidized nucleotide pools by hydrolyzing deoxyguanosine
triphosphate to deoxyguanosine monophosphate and thus prevents incorporation
of 8-oxoguanine into newly synthesized DNA. The second, OGG1,
encodes an enzyme that removes 8-oxoguanine from 8-oxoguanine�cytosine
pairs (Go:C) arising through oxidation of double-stranded
DNA. The third, MYH, encodes an enzyme that is responsible for
the removal of adenines from mispairs with 8-oxoguanine that arise
during replication of oxidized DNA. Although no mutations were
detected in the OGG1 and MTH genes, both alleles of the
MYH gene were shown to be mutated in the germ line of several
of the patients studied by Sieber et al. This finding is consistent
with the observed spectrums of APC mutations in their polyps,
since G:C
T:A), unlike the
germ-line mutations in familial adenomatous polyposis and attenuated
adenomatous polyposis coli. Since this type of transversion
represents a known "footprint" of oxidative DNA damage, the
authors decided to study three genes encoding polypeptides that are
necessary for efficient processing of this type of damage (Figure
1). The first, MTH, encodes a polypeptide that
"sanitizes" oxidized nucleotide pools by hydrolyzing deoxyguanosine
triphosphate to deoxyguanosine monophosphate and thus prevents incorporation
of 8-oxoguanine into newly synthesized DNA. The second, OGG1,
encodes an enzyme that removes 8-oxoguanine from 8-oxoguanine�cytosine
pairs (Go:C) arising through oxidation of double-stranded
DNA. The third, MYH, encodes an enzyme that is responsible for
the removal of adenines from mispairs with 8-oxoguanine that arise
during replication of oxidized DNA. Although no mutations were
detected in the OGG1 and MTH genes, both alleles of the
MYH gene were shown to be mutated in the germ line of several
of the patients studied by Sieber et al. This finding is consistent
with the observed spectrums of APC mutations in their polyps,
since G:C![]() T:A transversions arise
in 50 percent of progeny DNA after the replication of a template containing
Go:A mispairs (Figure 1).
T:A transversions arise
in 50 percent of progeny DNA after the replication of a template containing
Go:A mispairs (Figure 1).
|
Sieber and colleagues1 screened for germ-line MYH mutations in 152 patients with multiple colorectal adenomas (3 to 100 adenomas) and 107 patients with adenomatous polyposis (>100 adenomas) who did not carry germ-line mutations in APC. Although the frequency of heterozygosity at the MYH locus in the two study groups was similar to that among controls, biallelic germ-line MYH mutations were identified in 6 of 21 patients with 15 to 100 polyps (28.6 percent) and 8 of 107 probands with adenomatous polyposis (7.5 percent). Three of the latter eight patients also had extracolonic features frequently associated with familial adenomatous polyposis: duodenal adenomas or congenital hypertrophy of the retinal pigment epithelium. No biallelic germ-line MYH mutations were found in 131 patients with fewer than 15 adenomas. Family histories, when available, demonstrated an autosomal recessive pattern of inheritance: only siblings were affected.
The striking finding of all three studies1,6,7 is the identification of two mutational hot spots in the MYH gene. Of the 36 germ-line mutations identified in biallelic configurations in white European patients, 31 (86 percent) resulted in the amino acid substitutions Y165C and G382D, and 14 of 18 patients with these substitutions (78 percent) had either a homozygous configuration (Y165C�Y165C or G382D�G382D) or a compound heterozygous configuration (Y165C�G382D). Patients with origins on the Indian subcontinent carried different MYH variants (Y90X and E466X).7 The Y165C and G382D mutations affect amino acid residues that have been highly conserved throughout evolution and that attenuate the enzymatic activity of the equivalent protein in Escherichia coli.6 The other missense mutations either affect conserved amino acid residues or lie close to conserved regions encoding structural motifs of MYH. They are thus likely to be pathogenic, but this prediction must be substantiated by biochemical studies, in order to confirm that the complete loss of MYH function is necessary to induce the transformation process in epithelial cells.
The phenotype resulting from the lack of repair of oxidative damage
associated with the loss of MYH is the most likely cause of the G:C![]() T:A
somatic mutations detected in the APC gene.6
The MYH malfunction will undoubtedly lead to mutations in numerous
other genes, some of which might be important for homeostasis of
the epithelial cells in colonic crypts. However, since similar extracolonic
manifestations are observed in patients with polyposis who have
germ-line mutations in MYH and those with mutated APC, the
latter gene might be an obligate mutational target of MYH deficiency
in epithelial-cell transformation. In this respect, it would be
interesting to learn whether other organs and tissues are affected by
biallelic MYH mutations.
T:A
somatic mutations detected in the APC gene.6
The MYH malfunction will undoubtedly lead to mutations in numerous
other genes, some of which might be important for homeostasis of
the epithelial cells in colonic crypts. However, since similar extracolonic
manifestations are observed in patients with polyposis who have
germ-line mutations in MYH and those with mutated APC, the
latter gene might be an obligate mutational target of MYH deficiency
in epithelial-cell transformation. In this respect, it would be
interesting to learn whether other organs and tissues are affected by
biallelic MYH mutations.
What bearing do the present findings have on management of the disease? Multiple colorectal adenomas are removed endoscopically at the time of diagnosis whenever possible, but when endoscopic intervention is not feasible, colectomy is the normal course of action. This status quo could change. Because of the autosomal recessive nature of the mutations, it is not as easy to identify persons with biallelic MYH mutations as it is to identify persons with familial adenomatous polyposis. However, rapid, cost-effective, polymerase-chain-reaction�based screening for mutations in MYH hot spots in lymphocyte DNA from patients presenting with multiple polyps should permit the identification of many of these persons. It is likely that endoscopic surveillance of the siblings of these persons would lead to effective prevention of cancer, as has been the case with families with hereditary nonpolyposis colorectal cancer.8 Moreover, index patients with identified biallelic MYH mutations might not have to undergo total colectomy. Should future studies show that the number of polyps in patients with multiple colorectal adenomas linked to biallelic MYH mutations remains low, endoscopic intervention, or at least less drastic surgery, followed by regular endoscopic screening, might be sufficient to prevent the onset of cancer.
The identification of asymptomatic persons with biallelic MYH mutations in the general population is not realistic at this time. The frequency of the Y165C and G382D allelic variants among normal controls is reported to be about 1 percent.1,6,9 If these frequencies, as well as those of other mutant alleles, are confirmed in a much larger population with various ethnic origins, then about 1 in 10,000 persons has a biallelic MYH mutation. This frequency is too low to justify large-scale screening, especially since the prevalence of multiple colorectal adenomas and the penetrance of the MYH defect are unknown at this time. But this assessment may change with time.
Source Information
From the Institute of Medical Radiobiology, University of Zurich, Zurich, Switzerland.
References
- Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal
adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N
Engl J Med 2003;348:791-799.
[Abstract/Full Text] - Spirio L, Olschwang S, Groden J, et al. Alleles of the APC gene: an attenuated form of familial polyposis. Cell 1993;75:951-957. [ISI][Medline]
- Laken SJ, Petersen GM, Gruber SB, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet 1997;17:79-83. [ISI][Medline]
- Frayling IM, Beck NE, Ilyas M, et al. The APC variants I1307K
and E1317Q are associated with colorectal tumors, but not always with a
family history. Proc Natl Acad Sci U S A 1998;95:10722-10727.
[Abstract/Full Text] - Olschwang S, Tiret A, Laurent-Puig P, Muleris M, Parc R, Thomas G. Restriction of ocular fundus lesions to a specific subgroup of APC mutations in adenomatous polyposis coli patients. Cell 1993;75:959-968. [ISI][Medline]
- Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of
MYH associated with somatic G:C
 T:A
mutations in colorectal tumors. Nat Genet 2002;30:227-232.
[Medline]
T:A
mutations in colorectal tumors. Nat Genet 2002;30:227-232.
[Medline]
- Jones S, Emmerson P, Maynard J, et al. Biallelic germline
mutations in MYH predispose to multiple colorectal adenoma and somatic G:CT:A
mutations. Hum Mol Genet 2002;11:2961-2967.
[Abstract/Full Text] - Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000;118:829-834. [ISI][Medline]
- Shinmura K, Yamaguchi S, Saitoh T, Kohno T, Yokota J. Somatic mutations and single nucleotide polymorphisms of base excision repair genes involved in the repair of 8-hydroxyguanine in damaged DNA. Cancer Lett 2001;166:65-69. [Medline]