NEJM
Medical Progress
|
William M. Lee, M.D.
In the past five years, two drugs have been withdrawn from the market by the Food and Drug Administration (FDA) for causing severe liver injury, a potential danger that had not been fully recognized in the course of the preapproval clinical trials. Reports of adverse drug reactions of any type engender fear and skepticism in the public about the actions of the pharmaceutical industry and the FDA.1 Drug-induced hepatic injury is the most frequent reason cited for the withdrawal from the market of an approved drug, and it also accounts for more than 50 percent of the cases of acute liver failure in the United States today. More than 75 percent of cases of idiosyncratic drug reactions result in liver transplantation or death.2 Recent efforts by the National Institutes of Health and the FDA have been directed toward a better understanding of these occurrences in order to improve the outcomes.3,4 In this article, I will review the pathogenesis of drug-induced liver injury, the common adverse drug reactions that involve the liver, and the process of drug approval.
Hepatic Biotransformation
The liver, located between the absorptive surface of the gastrointestinal tract and drug targets throughout the body, is central to the metabolism of virtually every foreign substance. Most drugs and xenobiotics are lipophilic, enabling them to cross the membranes of intestinal cells. Drugs are rendered more hydrophilic by biochemical processes in the hepatocyte, yielding water-soluble products that are excreted in urine or bile.5 This hepatic biotransformation involves oxidative pathways, primarily by way of the cytochrome P-450 enzyme system.6 After further metabolic steps, which usually include conjugation to a glucuronide or a sulfate or glutathione, the hydrophilic product is exported into plasma or bile by transport proteins located on the hepatocyte membrane, and it is subsequently excreted by the kidney or the gastrointestinal tract.
Types of Drug Reactions
Most drugs cause liver injury infrequently. These reactions are considered idiosyncratic, occurring at therapeutic doses from 1 in every 1000 patients to 1 in every 100,000 patients, with a pattern that is consistent for each drug and for each drug class. Idiosyncratic reactions are characterized by a variable delay or latency period, ranging from 5 to 90 days from the initial ingestion of the drug, and are frequently fatal if the drug is continued once the reaction has begun. In contrast, with a drug such as isoniazid, mild injury may disappear despite continued use. Rechallenge is typically met with a more severe reaction regardless of whether the initial reaction was severe or mild. A few drugs such as acetaminophen injure hepatocytes in a dose-dependent fashion, so that the administered dose is a stronger determinant of the likelihood of a reaction than the host's metabolic constitution. In addition to the dose received, the patient's age,6 sex,7 and body-mass index affect metabolism and, therefore, outcomes, as do the simultaneous use of other foods and drugs and physiologic changes such as pregnancy and renal8 or liver disease.
For reasons that are unclear, women generally predominate among patients with drug-induced liver injury, as illustrated in a recent study in which women accounted for 79 percent of reactions due to acetaminophen and 73 percent of idiosyncratic drug reactions.2 Substances such as phenobarbital, phenytoin, ethanol, cigarette smoke, and grapefruit juice that induce hepatic enzymes may alter plasma drug levels; such alteration, in turn, can result in extrahepatic adverse effects (e.g., torsade de pointes and drug interactions).6,9 Enzyme inducers have a dynamic role in enhancing hepatotoxicity, as exemplified by the interaction of ethanol with acetaminophen (Figure 1), in which ethanol (the inducer) enhances liver injury.10 Substrate competition occurs when ethanol and acetaminophen are taken simultaneously; the combination actually decreases the speed of the metabolism of acetaminophen to its harmful byproduct. However, ethanol simultaneously slows the degradation of the 2E1 isoform of cytochrome P-450, thus increasing the availability of the enzyme and enhancing the formation of the toxic metabolite once ethanol is withdrawn.
|

Targets of Cell Injury
At least six mechanisms that primarily involve the hepatocyte produce liver injury, and the manner in which various intracellular organelles are affected defines the pattern of disease (Figure 2). If high-energy reactions involving cytochrome P-450 enzymes lead to covalent binding of drug to intracellular proteins, intracellular dysfunction is apparently produced that results in the loss of ionic gradients, a decline in ATP levels, and actin disruption, cell swelling, and cell rupture (Figure 2A).15,16 Drugs that affect transport proteins at the canalicular membrane can interrupt bile flow. Certain drugs, for example, bind to or disable the bile salt export protein. This process causes cholestasis; however, little cell injury occurs (Figure 2B).11 Genetic defects in transporters, as in the multidrug-resistance�associated protein 3, in combination with hormones may promote cholestasis during pregnancy or during treatment with estrogen-containing medications. In mixed forms of hepatic injury, the combined failure of canalicular pumps and other intracellular processes allows toxic bile acids to accumulate, causing secondary injury to hepatocytes. If cells of the bile ducts are injured, a likely outcome is protracted or permanent cholestasis, a disorder that has been termed the "vanishing bile duct syndrome."
|
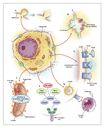
Drugs are relatively small molecules and, therefore, are unlikely to evoke an immune response. However, biotransformation involving high-energy reactions can result in the formation of adducts � that is, drugs covalently bound to enzymes. Adducts that are large enough to serve as immune targets may migrate to the surface of the hepatocyte, where they can induce the formation of antibodies (antibody-mediated cytotoxicity) or induce direct cytolytic T-cell responses (Figure 2C and Figure 2D). 12 The secondary cytokine response thus evoked may cause inflammation and additional neutrophil-mediated hepatotoxicity.17 Programmed cell death (apoptosis) can occur in concert with immune-mediated injury, destroying hepatocytes by way of the tumor necrosis factor (TNF) and the Fas pathways, with cell shrinkage and fragmentation of nuclear chromatin (Figure 2E).13 Proapoptotic receptor enzymes, if activated by drugs, will compete with protective so-called survival pathways within the cell, and this dynamic interaction may shift the balance either in favor of or against further cell damage.
Still other pathways to injury may develop when drugs damage mitochondria, disrupting fatty-acid oxidation and energy production. When drugs bind to or otherwise disable respiratory-chain enzymes or mitochondrial DNA, oxidative stress results, with ensuing anaerobic metabolism, lactic acidosis, and triglyceride accumulation (microvesicular fat within cells) (Figure 2F).14 Steatohepatitis (fat that primarily accumulates in the large vesicles outside the liver cells, with associated inflammation) is commonly associated with alcohol abuse, but it may also result from drugs.
Other cells within the liver may be the target of drug injury or serve as modulators of an incipient reaction. For example, Kupffer's cells activate cytokines that may amplify injury,18 and fat-storage cells (stellate cells) or macrophages may augment injury, produce fibrosis, or form granulomas. Chemotherapeutic agents can injure sinusoidal endothelial cells, a process that can lead to veno-occlusive disease.19 Therapeutic hormone administration may induce hepatocyte dedifferentiation, resulting in benign adenomas and, rarely, carcinomas.20 Clearly, multiple cellular pathways to liver injury are possible.
Pathogenesis
Most idiosyncratic drug reactions result from a succession of unlikely events, a "multihit" process. Genetic variants in isoenzymes that generate toxic byproducts are unlikely to cause severe toxicity alone, given that severe liver toxicity occurs so infrequently.5,21 In addition, hepatotoxicity may wax and wane with continuing drug use, implying that suppressor or attenuator pathways are active.17,22 Immune responses, once initiated, may be amplified or suppressed by means of the class I and class II major-histocompatibility-complex cell-surface receptors.12,17 The efficacy of drug-adduct peptide binding for antigen presentation depends on HLA configurations that have been genetically defined. For example, a specific HLA haplotype believed to be associated with hepatitis induced by the administration of amoxicillin�clavulanate was found in 57 percent of patients with the illness but in only 12 percent of unaffected persons.23
Cell-surface neoantigens may be short-lived, but they reappear with
continued exposure to the drug.24 Late events in
the immune sequence, such as expression of interleukin-10 or TNF-![]() ,
may augment or inhibit injury. For example, a specific interleukin-10
promoter phenotype that inhibits interleukin-10 secretion, which results
in down-regulation of type 2 helper T-cell immune reactions, is
linked to diclofenac toxicity.25 Variant TNF-
,
may augment or inhibit injury. For example, a specific interleukin-10
promoter phenotype that inhibits interleukin-10 secretion, which results
in down-regulation of type 2 helper T-cell immune reactions, is
linked to diclofenac toxicity.25 Variant TNF-![]() phenotypic expression has been implicated as a determining factor in
the severity of drug reactions related to acetaminophen.26
The xenobiotic constitutive androstane receptor has recently been
shown to be another key modulator of acetaminophen toxicity in mice
and potentially in humans, as well as a new target for
hepatoprotective strategies that is not mediated by immune responses.27
phenotypic expression has been implicated as a determining factor in
the severity of drug reactions related to acetaminophen.26
The xenobiotic constitutive androstane receptor has recently been
shown to be another key modulator of acetaminophen toxicity in mice
and potentially in humans, as well as a new target for
hepatoprotective strategies that is not mediated by immune responses.27
A series of events that first involve intracellular disruption, cell necrosis, or apoptosis, followed by activation of the immune sequence, might explain the features of idiosyncratic drug reactions: their rarity, their severity, and their resolution despite continued use of the drugs by patients with phenotypes that appear to be adaptive.22
It is possible, but not yet proven, that the genetic background of each patient could be addressed by pharmacogenomic approaches. Eventually, drug-mediated injuries may be prevented by screening methods that can identify aberrant gene polymorphisms or RNA-expression profiles before a patient uses a drug.21 Pharmacogenetic testing can identify unique cytochrome P-450 alleles that affect drug levels, but it is not clear whether specific markers of very rare idiosyncratic reactions can be identified, particularly if the reaction involves multiple steps.28
Clinical Consequences
The most frequent hepatotoxic drug reactions evoke moderate-to-severe injury to hepatocytes with a clinical picture that resembles viral hepatitis, characterized by a rapid onset of malaise and jaundice in association with elevated aminotransferase levels. Each drug has its own pattern of injury. If hepatocyte injury predominates, aminotransferase levels may be at least five times as high as normal. Elevations of alkaline phosphatase and bilirubin levels predominate in cholestatic syndromes. Signs of allergic reaction are absent in most patients. Acute liver failure may develop after a week or more of illness, particularly if the patient has continued the drug after the onset of symptoms. Death is not uncommon; elderly persons seem to be at particular risk, but specific data supporting this pattern are sparse.2
Idiosyncratic Reactions
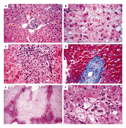
Idiosyncratic drug reactions made up 20 percent of cases of severe liver injury requiring hospitalization in a U.S. study involving 307 patients at six hospital centers (unpublished data). A variety of clinical patterns are observed (Table 1). The majority of idiosyncratic drug reactions involve damage to hepatocytes throughout the hepatic lobule, with various degrees of necrosis and apoptosis (Figure 3A and Figure 3B). Symptoms of hepatitis occur typically within days or weeks after the initial exposure and may continue to evolve even after the offending drug is withdrawn. Liver biopsy is seldom helpful for diagnosis.
|
|
Allergic Reactions
Some drug reactions have a striking allergic component. Sulfa drugs may induce fever, rash, and eosinophilia. Phenytoin is associated with fever, lymphadenopathy, rash, and severe hepatocyte injury � a group of signs that has been termed the "reactive metabolite syndrome" and that is slow to resolve in most instances.29 Halothane is also associated with this type of liver injury, though only after multiple exposures.30 The slow resolution of immunoallergic reactions suggests that allergens remain on the hepatocyte surface for weeks or months. Rapid recognition of toxic effects and immediate discontinuation of the offending agent are the keys to limiting hepatic and cutaneous damage. Even in the absence of systemic signs of allergy or peripheral-blood eosinophilia, eosinophilic infiltrates or granulomas may be present in a liver-biopsy specimen.
Bile-Duct Injury
When cholestasis predominates, injury to bile secretory components is present, either at the canalicular membrane or beyond (Figure 3C). Associated jaundice and pruritus may be severe, with permanent loss of bile ducts (Figure 3D).31
Dose-Related Acetaminophen Toxicity
Acetaminophen, as an example of a drug with dose-related toxic effects, rapidly causes hepatocyte injury, predominantly in the centrilobular region (Figure 3E). Acetaminophen toxicity produces the most common form of acute liver failure in the United States, accounting for 39 percent of cases in a recent survey of tertiary care centers.2 This type of liver injury occurs both after attempted suicide by acetaminophen overdose and after unintentional "therapeutic misadventures," in which use of the drug for pain relief in excess of the dose specified in the package labeling typically occurs over a period of several days.32 A careful medical history taking will clarify the quantity ingested; blood levels can be confirmatory but may not be elevated in cases of unintentional overdose. Extremely high aminotransferase values (typically exceeding 3500 IU per liter) help clinicians distinguish the toxic effects of acetaminophen from viral hepatitis or other drug injuries. N-acetylcysteine given for 36 to 72 hours reliably repletes glutathione and prevents injury if begun within 12 to 24 hours after ingestion. Cases of unintentional poisoning have a poorer outcome than suicide attempts. Chronic alcohol abuse, concomitant treatment with phenytoin and isoniazid, and starvation worsen liver injury related to acetaminophen.33,34 The incidence of acetaminophen poisoning and the severity of the outcomes vary widely throughout the world35; children are also occasionally susceptible to this form of acute liver failure.36 Changes in packaging that limit the number of doses and the accessibility of the drug may improve outcomes in all age groups.37 In addition to acetaminophen, other drugs also display a degree of dose-dependency in their capacity to cause liver injury (Table 2).
|
Less Common Drug Reactions
Several less frequent types of drug reactions involving the liver are included in Table 1. The rarer reactions typically involve nonparenchymal cells of the liver, such as sinusoidal endothelial cells.
Diagnosis and Treatment of Drug Reactions
It is difficult to identify a drug reaction with certainty. However, the possibility of a drug reaction must be considered in any patient with liver dysfunction. A careful drug history should be taken, which includes the patient's use of prescription, over-the-counter, herbal, or alternative medications. Injury induced by complementary and alternative medications has become more common as the use of these medications has increased.38,39 Various compounds � including germander, chaparral leaf, weight-loss preparations containing usnic acid,40 and many others � have been reported to be hepatotoxic. Other causes of liver dysfunction, such as viral hepatitis, hypotension, and biliary tract or liver disease related to alcohol abuse, must be excluded by a thorough medical history taking, ultrasonography, and appropriate serologic tests.
Assessment of causality can be difficult; often, many agents are used simultaneously, and questions about other potential causes may be inadvertently omitted. Causality-assessment methods provide a uniform approach to determine the likelihood of drug involvement in a suspected episode of hepatitis.41,42,43,44 Included among the standard factors that should be considered are the temporal relation (whether the onset of the symptoms was between 5 and 90 days after the initial exposure); the course after the patient stopped taking the drug (improvement within weeks); risk factors such as alcohol use, pregnancy, or old age; the concomitant use of drugs; the exclusion of causes other than drugs (e.g., viral hepatitis); patient's history with regard to previous toxic effects of the particular agent; and the response to rechallenge, if performed. In many instances, data are incomplete. Cautious rechallenge should be considered only if the diagnosis of drug-induced toxicity was highly questionable and only if no other drug is available to treat a serious problem.
Therapy for hepatotoxic effects of drugs consists of the immediate withdrawal of any and all suspected drugs. If a severe allergic reaction is observed, corticosteroids may be used, but no controlled trials have been performed to ascertain their efficacy. Similarly, ursodiol is frequently given for cholestatic liver injury, but it has not been subjected to careful study in this setting. Except for N-acetylcysteine for acetaminophen poisoning, there are no specific antidotes. The patient should be transferred to a liver-transplantation center if coagulopathy (as measured by an international normalized ratio of 1.5 or greater) or encephalopathy is present.
Hepatotoxicity in Patients with Chronic Liver Disease
Is the patient with liver disease more susceptible than others to liver injury? If liver function is impaired, one might expect a diminished likelihood of toxic reactions as a result of decreased enzyme activity. However, many enzyme systems are preserved, even in advanced liver disease, particularly those involved in conjugation reactions. In severe liver disease, the activity of the cytochrome P-450 2C19 isoenzyme is greatly decreased, whereas that of the 2D6 isoenzyme is intact.45,46 Increased levels of cytochrome P-450 2E1, as observed in nonalcoholic steatohepatitis, might enhance the toxicity of acetaminophen, but this effect has not been reported.47,48 Rates of drug metabolism in patients with cirrhosis may be reduced as much as 50 percent. Changes resulting from the increased fibrosis along the hepatic sinusoids in patients with cirrhosis further separate the bloodstream and hepatocyte.49
In general, patients with liver diseases are not uniformly at increased risk for hepatic injury, but there are some exceptions. Patients with hepatitis C do appear to be at increased risk for veno-occlusive disease after myeloablative therapy in preparation for bone marrow transplantation.50 Patients infected with the human immunodeficiency virus (HIV) who are being treated with highly active antiretroviral therapy may be at increased risk for hepatotoxic effects when they are coinfected with underlying liver diseases such as hepatitis B and C.51,52 The use of toxic drugs in the patient with established cirrhosis increases the risk of hepatic decompensation.53 Nonetheless, physicians cannot withhold vital medications from such patients. Thus, extra caution should be used in treating patients with underlying liver disease, because of potential for serious consequences. Frequent monitoring may be valuable but has not been shown to be cost effective and is often not performed. Since patients with cirrhosis are also prone to renal injury, aminoglycosides, radiocontrast agents, and prostaglandin inhibitors must be used with extreme caution in this group.
The Process of Drug Approval
Why is severe drug-induced liver injury often identified only after the drug is approved by the FDA? Each phase of clinical testing before approval includes close monitoring of serum liver-enzyme levels. Occasional increases in aminotransferase levels during clinical trials will not by themselves lead to the discontinuation of testing of a new drug, but the finding of frequent or more severe increases in aminotransferase levels (greater than eight times the upper limit of normal) or accompanying increases in bilirubin may do so. To detect a single case of clinically significant liver injury due to a drug with 95 percent confidence, the number of patients studied must be about three times the incidence of the reaction. A phase 3 study typically involves approximately 3000 patients. Idiosyncratic reactions can be expected to occur in less than 1 in 10,000 patients exposed; detecting a single reaction when its frequency is 1 in 10,000 would therefore require testing 30,000 patients. As a result, many drugs complete phase 3 testing and are approved before a case of idiosyncratic drug reaction is identified, since there is little chance of such a reaction in a small study cohort. Drugs that cause hepatotoxic effects will lead to acute liver failure in approximately 10 percent of those in whom drug-related jaundice develops.54 Thus, any drug that causes jaundice in preapproval trials will probably lead to acute liver failure when larger numbers of patients are exposed. Such drugs now require additional scrutiny before approval by the FDA.4
After approval, a much larger and more diverse group of patients is often exposed than was the case in the carefully controlled prelicensing trial. This wider range of patients, with doses, durations of treatment, and concomitant conditions beyond those encountered in the preapproval trials, will include several categories of patients at elevated risk for adverse effects: patients with renal failure or heart failure, patients with HIV infection or the acquired immunodeficiency syndrome, pregnant women, elderly persons, and children. After FDA approval, pharmaceutical companies are required to report serious adverse events (any incident resulting in death, a threat to life, hospitalization, or permanent disability) to the agency within 24 hours.55 Surveillance becomes a passive process once a drug is on the market, with most cases reported through the FDA's MedWatch program, through which physicians and pharmacists may voluntarily file written reports. It is estimated that MedWatch receives reports of fewer than 10 percent of adverse drug reactions.56 A recent study from France suggests that, at least in that country, fewer than 6 percent of hepatic adverse drug reactions are ever reported.57
Drugs Recently Withdrawn from the Market
Two drugs that were withdrawn because of hepatotoxicity � bromfenac and troglitazone � provide examples of problems encountered in the postapproval period. Bromfenac, a nonsteroidal anti-inflammatory drug marketed as Duract, was introduced in 1997 as a short-term analgesic for orthopedic pain.58 Nonsteroidal drugs, as a class, including the newer cyclooxygenase-2 inhibitors, have been associated with considerable hepatotoxicity.59,60,61 The FDA approved bromfenac for use for periods of 10 days or less, but longer periods of treatment were clearly possible after approval. Once released, bromfenac was associated with more than 50 cases of severe liver injury, and the drug was withdrawn in June 1998. All patients in whom toxicity was observed had been taking the drug for more than 30 days.62
Troglitazone (Rezulin) was the first of a new class of compounds, the thiazolidinediones, approved by the FDA in January 1997. A nuclear regulatory factor peroxisome proliferator-activated receptor gamma agonist, troglitazone reduces insulin resistance and increases insulin-stimulated glucose disposal, improving glucose control for patients with type 2 diabetes. In clinical trials, reversible elevations of serum aminotransferase levels were observed, occasionally exceeding eight times the upper limit of normal, but no examples of liver failure were identified. Once the drug was approved, however, reports of severe and fatal liver injury began to appear.63,64,65,66
The pathogenesis of troglitazone toxicity is not understood.67 Unlike bromfenac, troglitazone was not immediately removed from the market, because its benefits were initially thought to outweigh the risks. Over time, despite the addition of a black-box warning to the package insert that suggested monitoring of aminotransferase levels monthly, the number and severity of cases of hepatotoxic effects (a total of more than 90, of which at least 68 were fatal and 10 necessitated transplantation) prompted the FDA to withdraw troglitazone from the open market three years after its approval. A factor in the decision to withdraw the drug was the approval in May 1999 and July 1999 of two new thiazolidinediones, rosiglitazone (Avandia) and pioglitazone (Actos). These newer agents do not have the same degree of toxicity, although severe liver injury has been reported.68,69,70 Although there may be intrinsic differences in these newer drugs, more careful screening of patients by alert physicians, monitoring of aminotransferase levels, and early discontinuation of therapy in the event of moderately severe cases (as a result of the increased awareness) may contribute to the apparent greater safety of the newer drugs. Depending on the therapeutic importance of a drug, it may continue to be used despite its risks. Isoniazid71 remains a first-line agent against tuberculosis, even though increased levels of aminotransferase are observed in 15 to 20 percent of patients who take the medication and 1 in 1000 patients will have severe hepatic necrosis.72
These examples highlight the difficulties encountered by the FDA in providing oversight of the drug-testing process. It is impossible to predict every possible eventuality accurately from clinical trials. In reviewing new agents for approval, the FDA is charged with protecting the public from harm while recognizing each drug's predicted health benefits. However, it is only after a drug is approved and tested on the open market that its ultimate benefit and risk can truly be determined.
Approach to New Drugs
Case reports of toxic effects that appear in the first years after a drug is approved may help the FDA determine which new agents require closer scrutiny, as exemplified by some recent case reports of hepatotoxic effects of newer agents.73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100 Patients may not initially report taking complementary and alternative medications, which do not require prescriptions or FDA oversight. A careful inquiry by the physician may be necessary to identify the use of complementary medications and determine their role in liver injury.38,39,40,101,102,103 Monitoring aminotransferase levels on a monthly basis for the first six months of treatment has been suggested for patients taking medications that are known hepatotoxins, such as isoniazid or diclofenac. However, monitoring is unlikely to be effective in the case of a rare adverse reaction, such as that seen with terbinafine (incidence, 1 in 50,000). Monitoring is seldom performed consistently, and even if it were, it provides no guarantee of safeguarding the patient, since many drug reactions develop abruptly.104 Many deaths could be prevented if the offending drug were discontinued at the first sign of an adverse reaction.
Source Information
From the Division of Digestive and Liver Diseases, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas.
Address reprint requests to Dr. Lee at the Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390-9151, or at [email protected].
References
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug
reactions in hospitalized patients. JAMA 1998;279:1200-1205.
[Abstract/Full Text] - Ostapowicz G, Fontana RJ, Schi�dt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002;137:947-954. [ISI][Medline]
- Bissell DM, Gores GJ, Laskin DL, Hoofnagle JH. Drug-induced liver injury: mechanisms and test systems. Hepatology 2001;33:1009-1013. [CrossRef][ISI][Medline]
- Center for Drug Evaluation and Research. Drug-induced liver toxicity. (Accessed July 8, 2003, at http://www.fda.gov/cder/livertox/default.htm.)
- Weinshilboum R. Inheritance and drug response. N Engl J Med
2003;348:529-537.
[Full Text] - Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 2001;14:611-650. [ISI][Medline]
- Hunt CM, Westerkam WR, Stave GM. Effect of age and gender on the activity of human hepatic CYP3A. Biochem Pharmacol 1992;44:275-283. [ISI][Medline]
- Ikemoto S, Imaoka S, Hayahara N, Maekawa M, Funae Y. Expression of hepatic microsomal cytochrome P450s as altered by uremia. Biochem Pharmacol 1992;43:2407-2412. [CrossRef][ISI][Medline]
- Moss AJ. The QT interval and torsades de pointes. Drug Saf 1999;21:Suppl 1:5-10. [ISI][Medline]
- Thummel KE, Slattery JT, Ro H, et al. Ethanol and production of the hepatotoxic metabolite of acetaminophen in healthy adults. Clin Pharmacol Ther 2000;67:591-599. [ISI][Medline]
- Trauner M, Meier PJ, Boyer J. Molecular pathogenesis of
cholestasis. N Engl J Med 1998;339:1217-1227.
[Full Text] - Robin M-A, Le Roy M, Descatoire V, Pessayre D. Plasma membrane cytochromes P450 as neoantigens and autoimmune targets in drug-induced hepatitis. J Hepatol 1997;26:Suppl 1:23-30. [Medline]
- Reed JC. Apoptosis-regulating proteins as targets for drug discovery. Trends Mol Med 2001;7:314-319. [CrossRef][ISI][Medline]
- Pessayre D, Berson A, Fromenty B, Mansouri A. Mitochondria in steatohepatitis. Semin Liver Dis 2001;21:57-69. [CrossRef][ISI][Medline]
- Yun CH, Okerholm RA, Guengerich FP. Oxidation of the antihistamine drug terfenadine in human liver microsomes: role of cytochrome P-450 3A(4) in N-dealkylation and C-hydroxylation. Drug Metab Dispos 1993;21:403-409. [Abstract]
- Beaune P, Dansette PM, Mansuy D, et al. Human anti-endoplasmic reticulum autoantibodies appearing in a drug-induced hepatitis are directed against a human liver cytochrome P-450 that hydroxylates the drug. Proc Natl Acad Sci U S A 1987;84:551-555. [ISI][Medline]
- Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D,
Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci 2002;66:166-176.
[Abstract/Full Text] - Jonsson JR, Edwards-Smith CJ, Catania SC, et al. Expression of cytokines and factors modulating apoptosis by human sinusoidal lymphocytes. J Hepatol 2000;32:392-398. [CrossRef][ISI][Medline]
- DeLeve LD, Shulman H, McDonald GB. Toxic injury to hepatic sinusoids: sinusoidal obstruction syndrome (veno-occlusive disease). Semin Liver Dis 2002;22:27-42. [CrossRef][ISI][Medline]
- Brosens I, Johannisson E, Baulieu E-E, et al. Oral contraceptives and hepatocellular carcinoma. Br Med J 1986;292:1667-1668.
- Huang Y-S, Chern H-D, Su W-J, et al. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology 2002;35:883-889. [CrossRef][ISI][Medline]
- Watkins PB. Drug metabolism by cytochromes P450 in the liver and small bowel. Gastroenterol Clin North Am 1992;21:511-526. [ISI][Medline]
- Hautekeete ML, Horsmans Y, Van Waeyenberge C, et al. HLA association of amoxicillin-clavulanate-induced hepatitis. Gastroenterology 1999;117:1181-1186. [ISI][Medline]
- Cai H, Guengerich FP. Reaction of trichlorethylene oxide with proteins and DNA: instability of adducts and modulation of functions. Chem Res Toxicol 2001;14:54-61. [CrossRef][ISI][Medline]
- Aithal GP, Daly AK, Leathart J, Yuanneng CP, Day CP. Promoter polymorphisms of interleukin-10 (IL-10) and interleukin-4 (IL-4) predict the risk of diclofenac-induced hepatotoxicity. Gastroenterology 2000;118:Suppl 2:A977-A977. abstract.
- Bernal W, Donaldson P, Underhill J, Wendon J, Williams R. Tumor necrosis genomic polymorphisms and outcome of acetaminophen (paracetamol)-induced acute liver failure. J Hepatol 1998;29:53-59. [CrossRef][ISI][Medline]
- Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of
acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science
2002;298:422-424.
[Abstract/Full Text] - Phillips KA, Veenstra DL, Oren E, Lee JK, Sadee W. Potential
role of pharmacogenomics in reducing adverse drug reactions: a systematic
review. JAMA 2001;286:2270-2279.
[Abstract/Full Text] - Kleckner H, Yakulis V, Heller P. Severe hypersensitivity to diphenylhydantoin with circulating antibodies to the drug. Ann Intern Med 1975;83:522-523. [ISI][Medline]
- Kenna JG. Immunoallergic drug-induced hepatitis: lessons from halothene. J Hepatol 1997;26:Suppl 1:5-12. [Medline]
- Chitturi S, Farrell GC. Drug-induced cholestasis. Semin Gastrointest Dis 2001;12:113-124. [Medline]
- Schi�dt FV, Rochling FJ, Casey DL, Lee WM. Acetaminophen
toxicity in an urban county hospital. N Engl J Med 1997;337:1112-1117.
[Abstract/Full Text] - Schmidt LE, Dalhoff K, Poulsen HE. Acute versus chronic alcohol consumption in acetaminophen-induced hepatotoxicity. Hepatology 2002;35:876-882. [CrossRef][ISI][Medline]
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994;272:1845-1850. [Abstract]
- Wang K, Huang YS, Deng JF, et al. Characteristics and risk factors of acetaminophen-induced hepatitis in Taiwan. Zhonghua Yi Xue Za Zhi (Taipei) 1999;62:369-375. [Medline]
- Heubi J, Barbacci MB, Zimmerman HJ. Therapeutic misadventures with acetaminophen hepatotoxicity after multiple doses in children. J Pediatr 1998;132:22-27. [ISI][Medline]
- Moynihan R. FDA fails to reduce accessibility of paracetamol
despite 450 deaths a year. BMJ 2002;325:678-678.
[Full Text] - Chitturi S, Farrell GC. Herbal hepatoxicity: an expanding but poorly defined problem. J Gastroenterol Hepatol 2000;10:1093-1099. [CrossRef]
- Stedman C. Herbal hepatotoxicity. Semin Liver Dis 2002;22:195-206. [CrossRef][ISI][Medline]
- Favreau JT, Ryu ML, Braunstein G, et al. Severe hepatotoxicity associated with the dietary supplement LipoKinetix. Ann Intern Med 2002;136:590-595. [ISI][Medline]
- Danan G, Benichou C. Causality assessment of adverse reactions to drugs. I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol 1993;46:1323-1330. [ISI][Medline]
- Benichou C, Danan G, Flahault A. Causality assessment of adverse reactions to drugs. II. An original model for validation of drug causality assessment methods: case reports with positive rechallenge. J Clin Epidemiol 1993;46:1331-1336. [ISI][Medline]
- Lucena MI, Camargo R, Andrade RJ, Perez-Sanchez CJ, Sanchez De La Cuesta F. Comparison of two clinical scales for causality assessment in hepatotoxicity. Hepatology 2001;33:123-130. [CrossRef][ISI][Medline]
- Kaplowitz N. Causality assessment versus guilt-by-association in drug hepatotoxicity. Hepatology 2001;33:308-310. [CrossRef][ISI][Medline]
- George J, Murray K, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology 1995;21:120-128. [ISI][Medline]
- Adedoyin A, Arns PA, Richards WO, Wilkinson GR, Branch RA. Selective effect of liver disease on the activities of specific metabolizing enzymes: investigation of cytochromes P450 2C19 and 2D6. Clin Pharmacol Ther 1998;64:8-17. [ISI][Medline]
- Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 1998;27:128-133. [ISI][Medline]
- Burckart GJ, Frye RF, Kelly P, et al. Induction of CYP2E1 activity in liver transplant patients as measured by chlorzoxazone 6-hydroxylation. Clin Pharmacol Ther 1998;63:296-302. [ISI][Medline]
- Froomes PRA, Morgan DJ, Smallwood RA, Angus PW. Comparative effects of oxygen supplementation on theophylline and acetaminophen clearance in human cirrhosis. Gastroenterology 1999;116:915-920. [ISI][Medline]
- Strasser SI, Myerson D, Spurgeon CL, et al. Hepatitis C virus infection and bone marrow transplantation: a cohort study with 10-year follow-up. Hepatology 1999;29:1893-1899. [ISI][Medline]
- Clark CJ, Creighton S, Portmann B, Taylor C, Wendon JA, Cramp ME. Acute liver failure associated with antiretroviral treatment for HIV: a report of six cases. J Hepatol 2002;36:295-301. [CrossRef][ISI][Medline]
- Pol S, Vallet-Pichard A, Fontaine H. Hepatitis C and human immune deficiency coinfection at the era of highly active antiretroviral therapy. J Viral Hepat 2002;9:1-8. [CrossRef][ISI][Medline]
- Schenker S, Martin RR, Hoyumpa AM. Antecedent liver disease and drug toxicity. J Hepatol 1999;31:1098-1105. [CrossRef][ISI][Medline]
- Zimmerman HJ. Hepatotoxicity: the adverse effects of drugs and other chemicals on the liver. New York: Appleton-Century-Crofts, 1978.
- Code of federal regulations. 21. Good clinical practices. Princeton, N.J.: Bristol-Myers Squibb, 1996.
- Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM,
Bor DH. Timing of new black box warnings and withdrawals for prescription
medications. JAMA 2002;287:2215-2220.
[Abstract/Full Text] - Sgro C, Clinard F, Ouazir K, et al. Incidence of drug-induced hepatic injuries: a French population-based study. Hepatology 2002;36:451-455. [CrossRef][ISI][Medline]
- Meadows M. Serious liver injury: leading reason for drug removals, restrictions. FDA Consum 2001;35:8-9.
- Carrillo-Jimenez R, Nurnberger M. Celecoxib-induced acute
pancreatitis and hepatitis: a case report. Arch Intern Med 2000;160:553-554.
[Full Text] - Banks AT, Zimmerman HJ, Ishak KG, Harter JG. Diclofenac-associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 1995;22:820-827. [ISI][Medline]
- Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis 2002;22:169-183. [CrossRef][ISI][Medline]
- Fontana RJ, McCashland TM, Benner KG, et al. Acute liver failure associated with prolonged use of bromfenac leading to liver transplantation. Liver Transpl Surg 1999;5:480-484. [ISI][Medline]
- Gitlin N, Julie NL, Spurr CL, Lim KN, Juarbe HM. Two cases of severe clinical and histologic hepatotoxicity associated with troglitazone. Ann Intern Med 1998;129:36-38. [ISI][Medline]
- Neuschwander-Tetri BA, Isley WL, Oki JC, et al. Troglitazone-induced hepatic failure leading to liver transplantation: a case report. Ann Intern Med 1998;129:38-41. [ISI][Medline]
- Murphy EJ, Davern TJ, Shakil OA, et al. Troglitazone-induced fulminant hepatic failure. Dig Dis Sci 2000;45:549-553. [CrossRef][ISI][Medline]
- Malik AH, Prasad P, Saboorian MH, Thiele DH, Malet PF. Hepatic injury due to troglitazone. Dig Dis Sci 2000;45:210-214. [CrossRef][ISI][Medline]
- Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol 2000;13:135-140. [CrossRef][ISI][Medline]
- Al-Salman J, Arjomand H, Kemp DG, Mittal M. Hepatocellular injury in a patient receiving rosiglitazone: a case report. Ann Intern Med 2000;132:121-124. [ISI][Medline]
- Forman LM, Simmons DA, Diamond RH. Hepatic failure in a patient taking rosiglitazone. Ann Intern Med 2000;132:118-121. [ISI][Medline]
- May LD, Lefkowitch JH, Kram MT, Rubin DE. Mixed hepatocellular-cholestatic liver injury after pioglitazone therapy. Ann Intern Med 2002;136:449-452. [ISI][Medline]
- Garibaldi RA, Drusin RE, Ferebee SH, Gregg MB. Isoniazid associated hepatitis: report of an outbreak. Am Rev Respir Dis 1972;106:357-365. [ISI][Medline]
- Nolan CM, Goldberg SV, Buskin SE. Hepatotoxicity associated
with isoniazid preventive therapy: a 7-year survey from a public health
tuberculosis clinic. JAMA 1999;281:1014-1018.
[Abstract/Full Text] - Sreenarasimhaiah J, Shiels P, Lisker-Melman M. Multiorgan failure induced by atorvastatin. Am J Med 2002;113:348-349. [CrossRef][ISI][Medline]
- Kaufman KR. Carbamazepine, hepatotoxicity, organic solvents, and paints. Seizure 1999;8:250-252. [CrossRef][ISI][Medline]
- Baylor P, Williams K. Interstitial nephritis, thrombocytopenia, hepatitis, and elevated serum amylase levels in a patient receiving clarithromycin therapy. Clin Infect Dis 1999;29:1350-1351. [CrossRef][ISI][Medline]
- Gonzalez de la Puenta MA, Calderon E, Espinosa R, Rincon M, Varela JM. Fatal hepatotoxicity associated with enalapril. Ann Pharmacother 2001;35:1492-1492.
- Capella D, Bruguera M, Fugueras A, Laporte J. Fluoxetine-induced hepatitis: why is postmarketing surveillance needed? Eur J Clin Pharmacol 1999;55:545-546. [CrossRef][ISI][Medline]
- Kraus I, Vitezic D, Oguic R. Flutamide-associated acute hepatitis in advanced prostate cancer patients. Int J Clin Pharmacol Ther 2001;39:395-399. [ISI][Medline]
- Lasso-de-la-Vega MC, Zapater P, Such J, Perez-Mateo M, Horga JF. Gabapentin-induced hepatotoxicity. Am J Gastroenterol 2001;96:3460-3462. [CrossRef][ISI][Medline]
- Vergis E, Paterson DL, Singh N. Indinavir-associated hepatitis in patients with advanced HIV infection. Int J STD AIDS 1998;9:53-53. [CrossRef][ISI][Medline]
- Sphar L, Rubbia-Brandt L, Marinescu O, Armenian B, Hadengue A. Acute fatal hepatitis related to levofloxacin. J Hepatol 2001;35:308-309. [CrossRef][ISI][Medline]
- Bosch X. Losartan-induced hepatotoxicity. JAMA 1997;278:1572-1572. [CrossRef][ISI][Medline]
- Hwang I, Daniels AM, Holtzmuller KC. "Ecstasy"-induced hepatitis in an active duty soldier. Mil Med 2002;167:155-156. [ISI][Medline]
- Deltenre P, Berson A, Marcellin P, Degott C, Biour M, Pessayre
D. Mesalazine (5-aminosalicylic acid) induced chronic hepatitis. Gut
1999;44:886-888.
[Abstract/Full Text] - Babich MM, Pike I, Shiffman ML. Metformin-induced acute hepatitis. Am J Med 1998;104:490-492. [CrossRef][ISI][Medline]
- Piliero PJ, Purdy B. Nevirapine-induced hepatitis: a case series and review of the literature. AIDS Read 2001;11:379-382. [Medline]
- Aranda-Michel J, Koehler A, Bejarano PA, et al. Nefazodone-induced liver failure: report of three cases. Ann Intern Med 1999;130:285-288. [ISI][Medline]
- Romero-Gomez M, Suarez Garcia E, Fernandez MC. Norfloxacin-induced acute cholestatic hepatitis in a patient with alcoholic liver cirrhosis. Am J Gastroenterol 1999;94:2324-2325. [CrossRef][ISI][Medline]
- Benbow SJ, Gill G. Paroxetine and hepatotoxicity. BMJ
1997;314:1387-1387.
[Full Text] - Rosh JR, Dellert SF, Narkewicz M, Birnbaum A, Whitington G.
Four cases of severe hepatotoxicity associated with pemoline: possible
autoimmune pathogenesis. Pediatrics 1998;101:921-923.
[Full Text] - Hartleb M, Rymarczyk G, Januszewski K. Acute cholestatic hepatitis associated with pravastatin. Am J Gastroenterol 1999;94:1388-1390. [CrossRef][ISI][Medline]
- Ribeiro JM, Lucas M, Baptista A, Victorino RM. Fatal hepatitis with ranitidine. Am J Gastroenterol 2000;95:559-560. [CrossRef][ISI][Medline]
- Benazzi F. Risperidone-induced hepatotoxicity. Pharmacopsychiatry 1998;31:241-241. [ISI][Medline]
- Gupta AK, del Rosso JQ, Lynde CW. Brown GH, Shear NH. Hepatitis associated with terbinafine therapy: three case reports and a review of the literature. Clin Exp Dermatol 1998;23:64-67. [CrossRef][ISI][Medline]
- Iqbal M, Geonka P, Young MF, Thomas E, Borthwick TR. Ticlopidine-induced cholestatic hepatitis: report of three cases and review of the literature. Dig Dis Sci 1998;43:2223-2226. [ISI][Medline]
- Olanow CW. Tolcapone and hepatotoxic effects. Arch Neurol
2000;57:263-267.
[Abstract/Full Text] - Fernandes NF, Martin RR, Schenker S. Trazodone-induced hepatotoxicity: a case report with comments on drug-induced hepatotoxicity. Am J Gastroenterol 2000;95:532-535. [CrossRef][ISI][Medline]
- Lucena MI, Andrade RJ, Rodrigo L, et al. Trovafloxacin-induced acute hepatitis. Clin Infect Dis 2000;30:400-401. [CrossRef][ISI][Medline]
- Konig SA, Schenk M, Sick C, et al. Fatal liver failure associated with valproate therapy in a patient with Friedreich's ataxia: review of valproate hepatotoxicity in adults. Epilepsia 1999;40:1036-1040. [ISI][Medline]
- Cardona X, Avila A, Castellanos P. Venlaxafine-associated hepatitis. Ann Intern Med 2000;132:417-417.
- Benninger J, Schneider HT, Schuppan D, Kirchner T, Hahn EG. Acute hepatitis induced by greater celandine (Chelidonium majus). Gastroenterology 1999;117:1234-1237. [ISI][Medline]
- Campo JV, McNabb J, Perel JM, Mazariegos GV, Hasegawa SL, Reyes J. Kava-induced fulminant hepatic failure. J Am Acad Child Adolesc Psychiatry 2002;41:631-632. [ISI][Medline]
- Angell M, Kassirer JP. Alternative medicine -- the risks of
the untested and unregulated remedies. N Engl J Med 1998;339:839-841.
[Full Text] - Graham DJ, Green L, Senior JR. Hyperacute liver failure induced by troglitazone: what did we learn and what do we need to learn? Hepatology 2002;36:168A-168A. abstract.