NEJM
|
Addictions are relapsing, remitting lifelong illnesses that are notoriously difficult to treat. One year after they have stopped drinking, approximately one third of patients with alcoholism remain abstinent, one third have resumed drinking but not at their former level of consumption, and one third have relapsed completely. A defining problem with respect to the treatment of addiction is that we do not know how to restore the addicted brain to its former state, and many therapies � for example, methadone maintenance for opiate addiction � do not even attempt to do so. Because of the difficulty of treating addictions and a lack of understanding of the benefits that partially successful therapy convey to patients, their families, and the community, many addicts are never identified or treated.
Life is complex, but many events can be defined as stress, if they are viewed from the simplistic perspective of their effect on the brain. Behavioral, physiological, and molecular mechanisms help the body adapt to stress. Adaptations that return function to a previous set point may be thought of as homeostatic.1 However, adaptation may also result in new physiological set points outside the normal range. This phenomenon, which is referred to as allostasis, is defined as "homeostasis through change."2
The body functions through the coordinated activity of systems evolved over millennia. Although humans are highly resilient and capable of surviving in the most stressful environments, it is not surprising that chronic or repeated stress increases the risk of a variety of diseases, including psychiatric disorders.
The addictions are among the more important sources of stress at the individual, family, and community levels. Recently, allostatic alteration of brain function through stress-related mechanisms has been identified as one component of the pathway to addiction.3 The brain survives addiction, but in the absence of the drug, the brain does not return to the base-line set point, and chronic dysphoria and anxiety are present. The clinical goal is to relieve the patient's depression and anxiety, but treatment of depression alone rather than in combination with treatment of addiction is often ineffective. Patients with alcoholism who are abstinent may nevertheless have extended periods of anxiety, depression, and sleep disturbances, predisposing them to relapse.
Levels of corticosteroids and corticotropin-releasing hormone are dramatically elevated after exposure to ethanol and are even higher during the initial period of withdrawal.4 After months of abstinence, however, the spinal fluid level of corticotropin-releasing hormone, which is derived primarily from extrahypothalamic sources, decreases. In rodents, behavioral and physiological responses to intracerebroventricular corticotropin-releasing hormone are increased during protracted withdrawal from ethanol, suggesting that the release of corticotropin-releasing hormone decreases and thus that corticotropin-releasing hormone receptors are up-regulated. Therefore, the molecular players in the physiological response to stress are potential therapeutic targets that may restore brain function to its preaddicted state.3
Mouse models in which genes have been disrupted or modified (Figure 1) are being used to refine our understanding of the genes, molecules, and circuits that link stress to drug-seeking behavior.4 In mice in which the gene for either of the two corticotropin-releasing hormone receptors � crhr1 and crhr2 � has been disabled, stress-induced activation of the hypothalamic�pituitary�adrenal axis is attenuated. This reduction in response is additive when the genes for both receptors are deleted. However, crhr1-knockout and crhr2-knockout mice are behaviorally different. Whereas crhr1-knockout mice are less anxious than wild-type mice, crhr2-knockout mice appear to be more anxious. Sillaber et al. found that mice lacking functional receptors for corticotropin-releasing hormone 1 (as a result of the deletion of the G-protein�coupling domain) consumed increasing amounts of ethanol after repeated exposure to stress in the form of frustration in social situations and forced swimming.5 At base line, however, fluid intake and alcohol consumption were the same in these mice and control mice. These effects appeared to be long-lasting, since they were present even nine months after exposure.
|
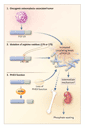
Accompanying the changes in behavior are base-line alterations in receptors and peptides that may be critical to the stress-induced drug preference of crhr1-knockout mice and to anxiety in crhr2-knockout mice. The NR2B glutamate receptor is up-regulated in both the hippocampus and the amygdala of crhr1-knockout mice. Because it is modulated by ethanol, NR2B could have a role in the mechanism of stress-induced alcohol preference in these animals. In addition, crhr1-knockout mice have increased hypothalamic levels of corticotropin-releasing hormone and vasopressin, another corticotropin secretagogue, presumably in order to maintain the function of the limbic�hypothalamic�pituitary�adrenal axis. Pituitary levels of vasopressin are also increased in crhr2-knockout mice. The increase in vasopressin could mediate the responsiveness of these mice to stressful stimuli, making them appear more anxious. Results of other studies indicate that vasopressin-mediated effects are critical in the disturbance in the function of the limbic�hypothalamic�pituitary�adrenal axis in both humans with depression and rats with anxiety.
These recent findings regarding stress, addiction, and alcohol preference
in normal and knockout mice indicate that a variety of systems and
their components, including N-methyl-D-aspartate and
vasopressin, which are affected by the dysregulation of the
limbic�hypothalamic�pituitary�adrenal axis, are potential
therapeutic targets in patients with alcoholism and other addictions.
Other systems that are known to be influenced by stress or alcohol,
including the noradrenergic, cholinergic, and dopaminergic systems
(which mediate stress- and drug-induced neuroadaptations), as well as
![]() -aminobutyric acid, opioids, and
neuropeptide Y (molecules that respond to neuroadaptation) are other
potential targets for therapy. Neuropeptide Y influences ethanol
preference, and a relatively common functional polymorphism of
neuropeptide Y � the substitution of proline for leucine at
position 7 (Leu7Pro) � has been reported to influence alcohol
consumption in humans. Several antidepressants decrease the levels of
corticotropin-releasing hormone, and corticotropin-releasing hormone�receptor
antagonists are being evaluated in both rodent and nonhuman primate
models of anxiety and substance abuse. Although the crhr1-knockout
mice studied by Sillaber et al. increased their intake of alcohol in
response to stress,5 it is important to
remember that the disruption of this receptor would have altered the
ontogeny of the limbic�hypothalamic�pituitary�adrenal axis.
Both disruption and overactivity of the stress axis may increase the
body's vulnerability to alcoholism. Learning more about the way in
which the brain changes in response to chronic stress and long-term
drug use should enable us to devise better therapies for alcoholism
and other stress-related neuropsychiatric disorders.
-aminobutyric acid, opioids, and
neuropeptide Y (molecules that respond to neuroadaptation) are other
potential targets for therapy. Neuropeptide Y influences ethanol
preference, and a relatively common functional polymorphism of
neuropeptide Y � the substitution of proline for leucine at
position 7 (Leu7Pro) � has been reported to influence alcohol
consumption in humans. Several antidepressants decrease the levels of
corticotropin-releasing hormone, and corticotropin-releasing hormone�receptor
antagonists are being evaluated in both rodent and nonhuman primate
models of anxiety and substance abuse. Although the crhr1-knockout
mice studied by Sillaber et al. increased their intake of alcohol in
response to stress,5 it is important to
remember that the disruption of this receptor would have altered the
ontogeny of the limbic�hypothalamic�pituitary�adrenal axis.
Both disruption and overactivity of the stress axis may increase the
body's vulnerability to alcoholism. Learning more about the way in
which the brain changes in response to chronic stress and long-term
drug use should enable us to devise better therapies for alcoholism
and other stress-related neuropsychiatric disorders.
David Goldman, M.D.
Christina S. Barr, V.M.D., Ph.D.
National Institute on Alcohol Abuse and Alcoholism
Rockville, MD 20852
References
- Selye H. The stress of life. New York: McGraw-Hill, 1976.
- McEwen BS. Stress, adaptation, and disease: allostasis and allostatic load. Ann N Y Acad Sci 1998;840:33-44. [Abstract/Full Text]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 2000;24:97-129. [ISI]
- Adinoff B, Martin PR, Bone GH, et al. Hypothalamic-pituitary-adrenal axis functioning and cerebrospinal fluid corticotropin releasing hormone and corticotropin levels in alcoholics after recent and long-term abstinence. Arch Gen Psychiatry 1990;47:325-330. [ISI][Medline]
- Sillaber I, Rammes G, Zimmermann S, et al. Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRH1 receptors. Science 2002;296:931-933. [Abstract/Full Text]